As a universal redox catalyst, iron is an essential micronutrient for all forms of life. It participates in various critical biochemical processes such as oxygen transport in hemoglobin and electron transport in cellular respiration.[1] Given its significance to both the host and the evading microbes, iron holds a pivotal position at the host-pathogen interface.[2] Many animals, in response to the invasion of extracellular pathogens, have evolved “nutritional immunity,” by which their cells, namely macrophages, sequester essential molecules, such as iron.[1,2,3] This hoarding strategy not only starves extracellular pathogens but also generates toxic reactive oxygen species that would kill intracellular bacteria.[3] However, some intracellular pathogens may have co-evolved to resist these toxic radicals and deploy varied mechanisms to acquire iron from the hosts to overcome nutritional immunity. Furthermore, the natural host defense mechanism may potentially lead to development of anemia of inflammation (AI) as a side effect of lowering serum iron levels.[3] Elucidation of the dynamics of iron metabolism in both the host macrophages and pathogens during bacterial infections may lay the foundation for future development of more effective antibiotics, AI treatments, or tissue iron overload.
In efforts to shed light on the mechanisms by which the host’s macrophages achieve nutritional immunity against Salmonella typhimurium (ST), Willemetz et al. investigated iron homeostasis in three mouse models presenting different iron levels in tissue macrophage. A/J WT mice have basal iron level, while pyruvate kinase-deficient AcB61 mice exhibit high iron level and Hepcidin knockout, or Hamp–/–, exhibit low iron level in their macrophages (Figure 1, top). Hamp–/– mice, though, have high iron levels systemically in the absence of hepcidin that normally would signal for the endocytosis of cellular iron exporter ferroportin (FPN) (Figure 1, bottom right panel). Over the course of ST infection, in Hamp–/– mice observed decrease in hematocrit percentage in the blood (Figure 1, bottom) as well as iron accumulation in the liver and the spleen. Serum iron level, on the other hand, increased while that of the WT increased. Altogether, these data suggest upregulation of FPN in infected Hamp–/– mice. However, Fpn mRNA expression was significantly suppressed in both the WT and the knockout mice. Even though this downregulation of FPN contradicts with all other measurements, this shows FPN regulation is independent of Hepcidin, the iron master regulator, during ST infection. Similar blood iron profile and the contradictory FPN protein level were observed in AcB61 mice (Figure 1, middle). The upregulation of heme oxygenase 1 (Hmox1), coupled with the presence of ingested red blood cells inside iron-overloaded hapatic and spleenic macrophages, suggest the high iron level may be results of high erythrophagocytosis activity (not shown). Fpn and Hamp mRNA expression level, meanwhile, remained constant over the course of the infection. This once again pointed to the independence of hepcidin in FPN regulation during ST infection.[3]

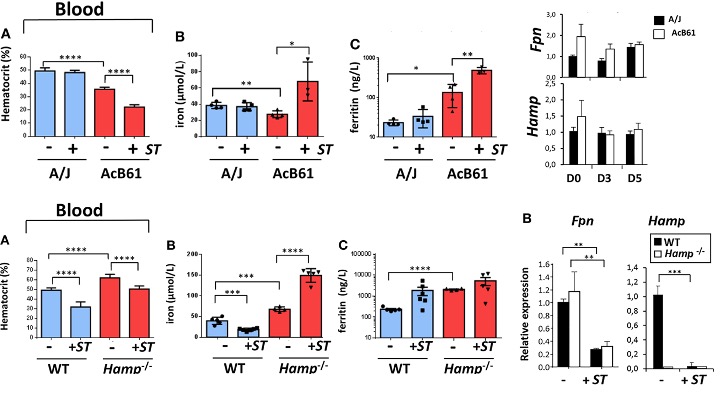
Figure 1. Serum profiles and Fpn and Hepcidin mRNA levels of three infected mice models.[3]
NLRP6 inflammasome is responsible for the downregulation of ferroportin during ST infection. NLRP6 inflamasome is a Nod-like receptor that induces the production of interleukin 18 upon the invasion of viral and extracellular gram-positive bacteria.[4] While NLRP6 is helpful in fighting against extracellular pathogens, Deng et als. found Nlrp6–/– mice infected with ST to have significantly higher survival rate and lower bacterial load systematically than the infected WT. Interestingly, while the infected WT mice experienced dysregulation of iron metabolism, infected Nlrp6–/– mice resist iron accumulation in the spleen as well as hypoferremia, evident by the higher serum and lower spleenic iron content than the WT three days after infection. Altogether, this suggests that NLRP6 is detrimental to the host control of ST infection and the maintenance of iron homeostasis. Further gene expression comparison between infected WT and Nlrp6–/– bone marrow-derived macrophages (BMDMs) suggest that NLRP6 regulates the host iron homeostasis by suppressing the FPN gene activator NRF2 post-transcriptionally. This leads to downregulation of the Fpn gene and the subsequent dysregulated iron metabolism in ST-infected WT mice.[2]

Figure 2. Regulatory mechanism of FPN-gene activator NRF2 by NLRP6.[2]
NLRP6 regulates NRF2 through interaction with AKT kinase. NLPR6 impedes AKT phosphorylation, which in turn curbs the activation of the mammalian target of rapamycin 1 (mTORC1) and FOXO3A transcription factor. The former protein is responsible for phosphorylating p62, which when binds to NRF2-regulating protein KEAP1 will allow NRF2 to dissociate from KEAP1 and translocate from the cytoplasm to the nucleus to express Fpn. In the absence of phosphorylated p62, KEAP1 will subject the bound NRF2 to ubiquitin-mediated degradation. FOXO3A transcription factor, when unphosphorylated, is an activator of Keap1. Upon phosphorylation by AKT, however, FOXO3A will be degraded. Therefore, hindrance of AKT phosphorylation saves FOXO3A from degradation and thereby increases the expression of KEAP1. Ultimately, the reduction of phosphorylated p62 to free NRF2 from elevating KEAP1 “hunters” obstructs NRF2 translocation to the nucleus and the expression of FPN (Figure 2).
Even though inconsistencies in the anemia symptoms and Fpn expression levels in both Willemetz et al. and Deng et al.’s work may point to the complexity of iron homeostasis in Salmonella typhimurium infection, their new insights on iron metabolism regulation mechanism by not hepcidin but NLRP6 inflammasome laid a solid foundation for further exploration of the host nutritional immunity against the extracellular pathogen.
References:
- Núñez G, Sakamoto K, Soares MP. Innate Nutritional Immunity. J Immunol. 2018 Jul 1;201(1):11-18. doi: 10.4049/jimmunol.1800325. PMID: 29914937; PMCID: PMC6028930.
- Deng Q, Yang S, Sun L, Huang K, Dong K, Zhu Y, Cao Y, Li Y, Wu S, Huang R. A detrimental role of NLRP6 in host iron metabolism during Salmonella infection. Redox Biol. 2022 Feb;49:102217. doi: 10.1016/j.redox.2021.102217. Epub 2021 Dec 18. PMID: 34942528; PMCID: PMC8695358.
- Willemetz A, Beatty S, Richer E, Rubio A, Auriac A, Milkereit RJ, Thibaudeau O, Vaulont S, Malo D, Canonne-Hergaux F. Iron- and Hepcidin-Independent Downregulation of the Iron Exporter Ferroportin in Macrophages during Salmonella Infection. Front Immunol. 2017 May 1;8:498. doi: 10.3389/fimmu.2017.00498. PMID: 28507548; PMCID: PMC5410627.
- Levy M, Shapiro H, Thaiss CA, Elinav E. NLRP6: A Multifaceted Innate Immune Sensor. Trends Immunol. 2017 Apr;38(4):248-260. doi: 10.1016/j.it.2017.01.001. Epub 2017 Feb 15. PMID: 28214100.