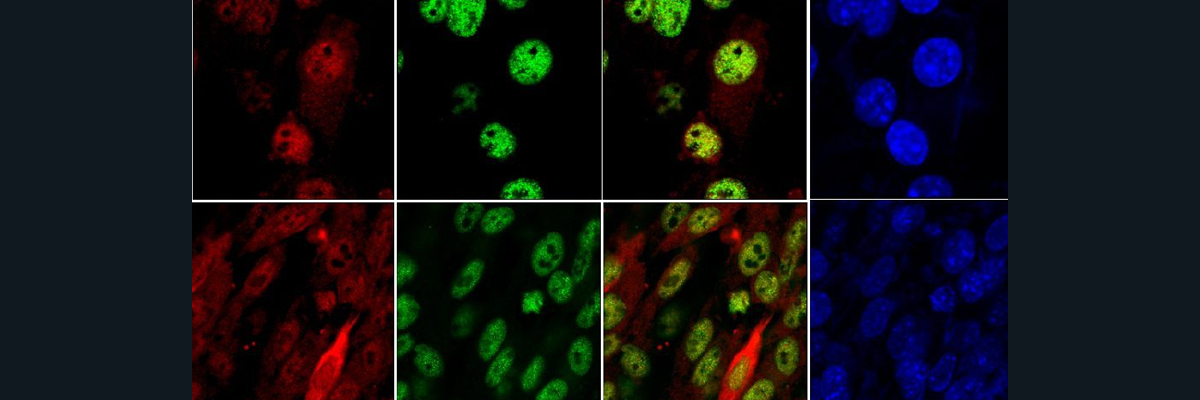
Copper is an essential metal that we get from daily intake of foods and water. Without it, certain copper-dependent enzymes (“cuproenzymes”) cease to function which has negative consequences in energy production, brain and muscle development, and other biochemical/physiological processes in our bodies [1]. Menkes and Wilson are genetic conditions associated with deficient and toxic levels of copper, respectively. The molecular cause is related to genetic mutations in two DNA protein-encoding genes: ATP7A and ATP7B. Under the same protein family as Na+/K+ pump, ATP7A/B transporters are critical for regulating copper concentrations inside and outside cells.
In an effort to develop possible drug-targeting therapies for addressing Menkes and Wilson, researchers have sought to determine the three-dimensional structure of ATP7A/B to better understand the mechanism behind how copper is transported and how mutations in the structure of the proteins influence the function. Unfortunately, there are no solved structures of ATP7A/B in the Protein Data Bank (PDB) or any other protein repositories as of this blog.
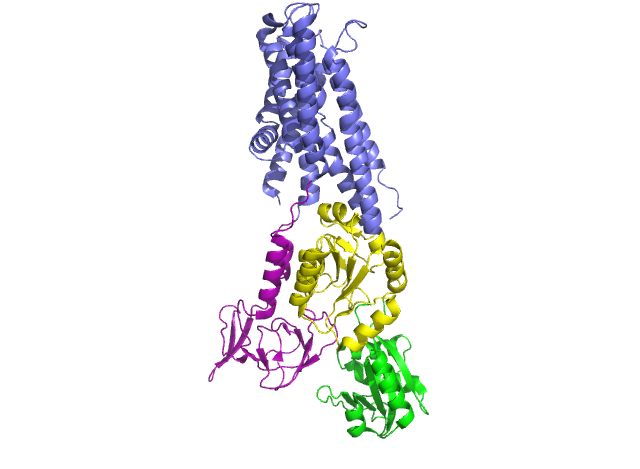
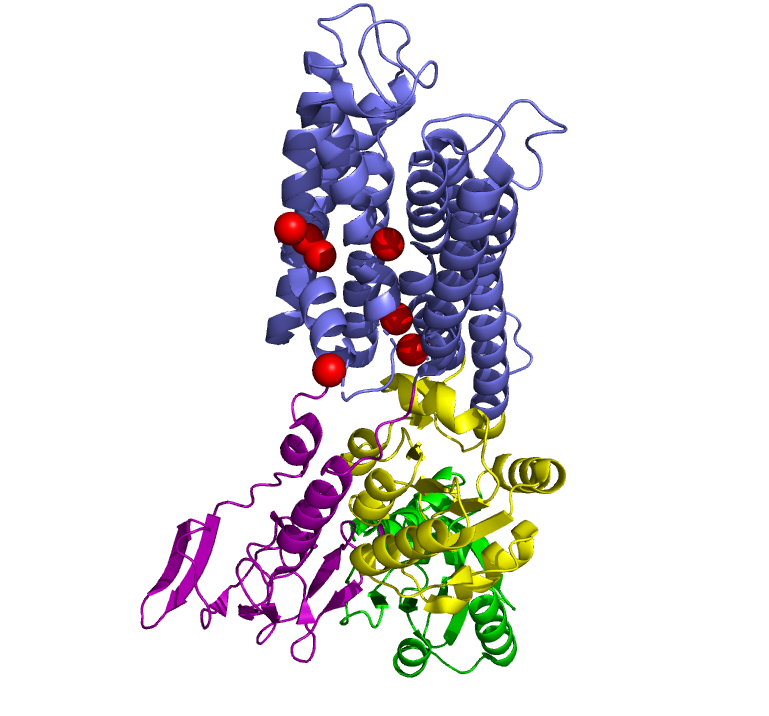
In 2011, Pontus Gourdon and Xiang-Yu Liu and other members of the Danish National Research Foundation sought to solve the structure of a bacterial Cu+-ATPase in Legionella pneumophilia, which is called “LpCopA” (CopA for short) [1]. In this journal article, the researchers were successfully able to extract, stabilize and determine the atomic structure of the copper transporter. For the most part, the structure of the protein contained these features: 8 transmembrane helices, two of which are docking platforms for a chaperone protein (which carries copper), CopZ. [2] Additionally, the structure also reveals the spatial arrangement of 3 important domains: nucleotide-binding domain (N; green), phosphorylating domain (P;yellow) and actuator domain (A;magenta).
When closely examining the amino acid composition of docking platform, it is hypothesized that the conserved residues K142, K135, and R136 electrostatically interact to the negatively charged interface of CopZ. Upon protein-protein interaction on the MB helix, the copper ion is released and interacts with three residues: D337, M148 and E205. Once ATP binds to the N domain, autophosphorylation occurs in the P domain, leading to a conformational change, allowing the copper to move deeper into the membrane. Two conserved residues guide the copper inside the membrane: C382 and C384. Just before dephosphorylation, the copper is then able to move to the final site, where it exits to the other side of the membrane.
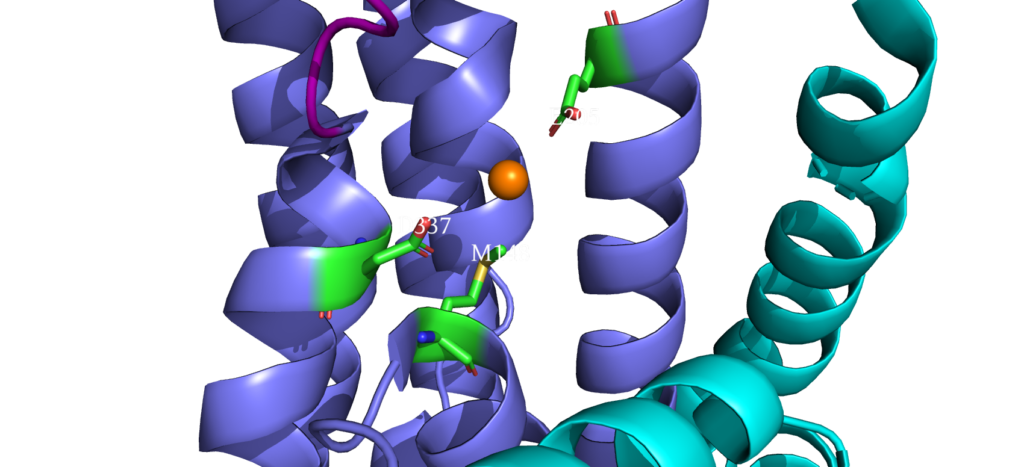
Copper Entry Site 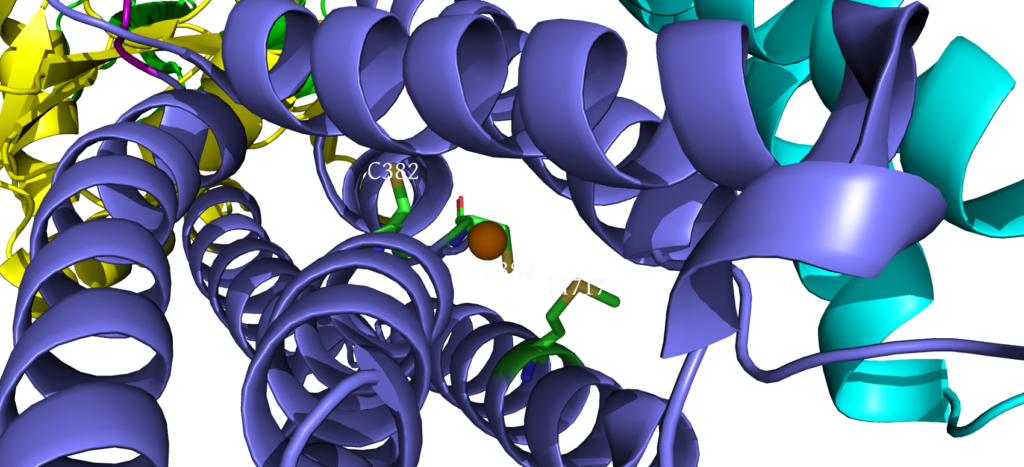
Cysteine Residues interacting with Copper 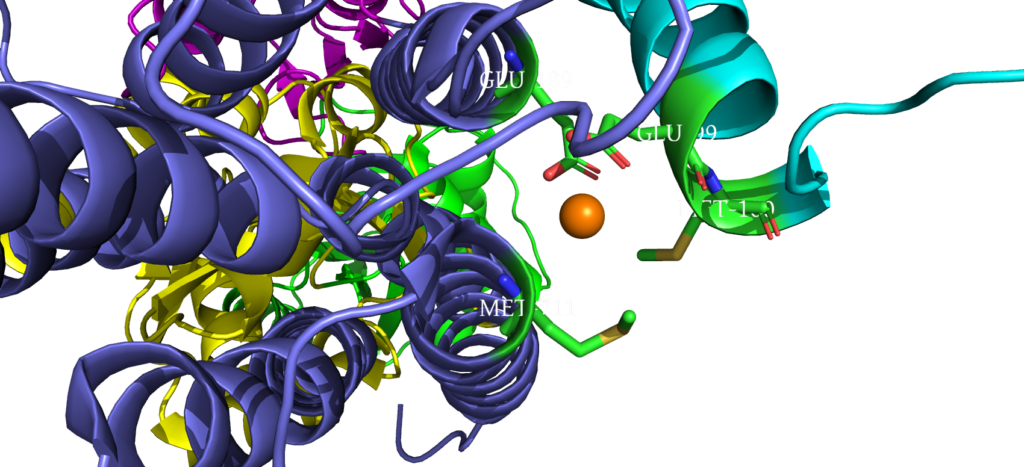
Copper Exit Site
When comparing the amino acid sequences of CopA with Human ATP7A/B, there was about a ~35-36% similarity in conserved residues, which met the criteria for being able to 1) use the bacterial protein to model Menkes/Wilson and 2) create homology models of ATP7A/B [3]. For the former, the researchers found that by overlaying missense mutations in ATP7A/B on the CopA protein, most of the perturbations were connected to impacting functionally important regions (N and P, for example). For the latter, the homology models were derived roughly by using CopA as a baseline, combined with experimental and computational input. Creating these models enables scientists to better understand the molecular pathophysiology of Menkes/Wilson and how severity is dependent on localization of these mutant variants.
This article was written by Justin Nguyen (contact info: jnguyen01@wesleyan.edu).
References:
1. Gourdon, P. et al. Crystal structure of a copper-transporting PIB-type ATPase. Nature 475, 59–64 (2011).
2. Argüello, J. M., Raimunda, D. & Padilla-Benavides, T. Mechanisms of copper homeostasis in bacteria. Front. Cell. Infect. Microbiol. 3, (2013).
3. Gourdon, P., Sitsel, O., Karlsen, J. L., Møller, L. B. & Nissen, P. Structural models of the human copper P-type ATPases ATP7A and ATP7B. Biological Chemistry 393, 205–216 (2012).