Iron is an essential metal that plays several key roles in the body. Most importantly, it helps transport oxygen by being a crucial part of hemoglobin, the protein in red blood cells that carries oxygen throughout the body. Iron is also important for a healthy immune system [1,2]. Iron levels in the body are tightly regulated through a network of proteins, enzymes, and transporters, including transferrin, ferroportin, hepcidin, ZIP14, ZIP8, and the HFE protein [1,3]. Transferrin carries iron in an inactive form (Fe³⁺) to cells by binding to a receptor (TfR1) on the cell surface, where the iron is then converted into its active form (Fe²⁺) and used by the cell [1]. HFE protein helps limit iron intake by blocking transferrin’s ability to deliver iron when levels are high [4]. Ferroportin is the body’s main iron “exit” pump, releasing iron from cells to the bloodstream. Hepcidin stops this release when blood iron levels are high, preventing overload. In addition to the previously mentioned transports, recently, scientists discovered that ZIP8 and ZIP14—proteins previously known for transporting zinc—also help move iron from the blood into cells. This is an exciting finding because it reveals that ZIP8 and ZIP14 contribute to iron transport as well. When there is too much iron in the blood, transferrin becomes saturated, and ZIP8 and ZIP14 provide an alternative way for cells to absorb iron that is not bound to the transferrin [1]. When iron levels aren’t well-regulated, serious health issues can occur. Low iron levels can lead to anemia, while too much iron can cause hemochromatosis, a genetic disorder where iron builds up in organs like the liver, pancreas, and heart. Hemochromatosis is divided into two types based on the severity and the causes. Type 1 hemochromatosis, is caused by mutations in the HFE gene, such as C282Y, which disrupts the folding of the HFE protein and prevents it from binding to the transferrin receptor, allowing unchecked iron absorption and accumulation in cells, particularly in the liver [5]. Type 2 hemochromatosis is caused by mutations in the HFE2 gene. The HFE2 mutation leads to reduced hepcidin levels. Low hepcidin allows ferroportin to continuously transport iron into the bloodstream, causing excess iron. This overload saturates transferrin, leading to Non-Transferrin Bound Iron (NTBI), which cells then absorb, further increasing iron accumulation. This condition results in rapid and severe iron buildup, especially in the liver, and also affects the pancreas and heart.
People with hemochromatosis (whether type 1 or 2) experience symptoms such as white, flat nails, joint pain, dry skin, and darkened skin patches (melanoderma). Additionally, they are at higher risk for serious problems and diseases like heart failure and liver cancer (hepatocellular carcinoma) [6]. Until about 2014, scientists mainly focused on how iron bound to transferrin contributed to hemochromatosis, often overlooking the role of NTBI. In 2015, Dr. Mitchell D. Knutson from the University of Florida published a study that highlighted ZIP14, a protein that helps cells absorb NTBI, as a potential factor in iron overload in hemochromatosis. To test this, researchers use different mice models- male and female. The researchers started off by initially injecting mice with ferric citrate in order to saturate the plasma and thus the transferrin with iron. This injection was followed by a second injection that had 59 Fe-labeled ferric citrate (radioactive isotope that emits gamma radiation), and this would represent the NTBI. They used this radioactive isotope because they wanted to track where this NTBI was taken up by the various tissues that they examined which were the liver, pancreas, kidney, heart, and spleen. After two hours, the mice were sacrificed, and the tissue was collected for gamma counting (Figure 1A). Scientists found that NTBI uptake in the liver and the pancreas of the mice that had ZIP14, encoded by Slc39a14 gene, knocked out ( Slc39a14–/– Mice: meaning that ZIP14 is no longer expressed) was nearly 70% lower compared to the healthy mice expressing ZIP14 (WT mice). On the other hand, NTBI was higher in the kidney, heart, spleen, and carcass of the ZIP14 knockout mice. This suggests that the NTBI, which could not be absorbed by the liver and pancreas, is instead taken up by the heart, kidneys, and spleen in a ZIP14-independent manner. Additionally, scientists confirmed their knock-out process in all of the models by performing a western blot (an experiment that shows if the protein is expressed or not using protein-specific antibodies- Figure 1B). They also looked at heterozygous mice, which had about 50% of the normal SLC39A14 protein levels. They discovered that even with just one functional copy of the gene, these mice showed normal NTBI clearance in the liver (Figure 1C). To dig deeper into whether the impaired NTBI uptake was specifically happening in hepatocytes (the main liver cells), they isolated primary hepatocytes from the mice. They found that NTBI uptake in these cells was also significantly reduced in the knockout mice, just like it was in the liver (Figure 1D). Now, the team knew that SLC39A14 was involved in NTBI uptake, but they also wanted to see if it affected the uptake of transferrin-bound iron (TBI). Interestingly, they found that TBI uptake was not altered in the liver, pancreas, heart, kidney, or spleen of the knockout mice, indicating that SLC39A14 is not necessary for TBI uptake in these tissues (Figure 1E)
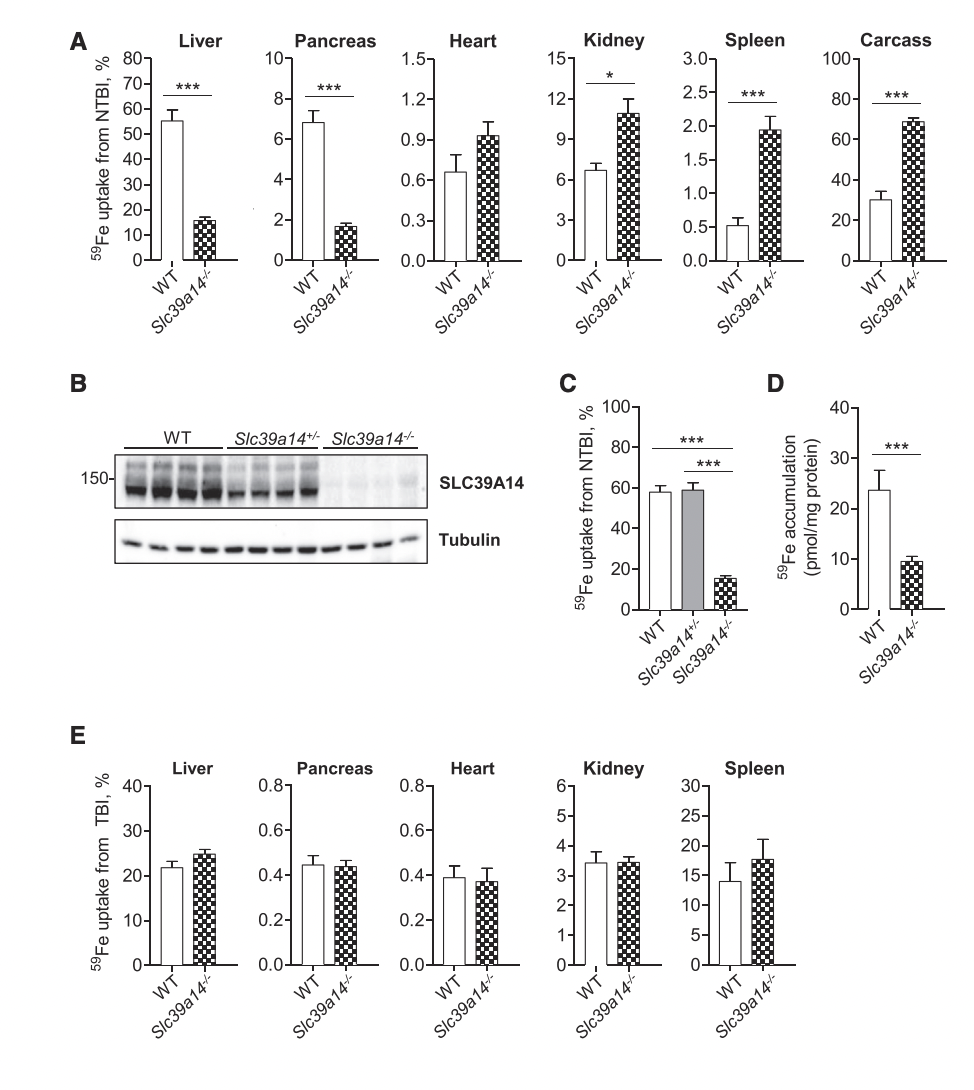
Figure 1
After that, scientists moved to test what happens to NTBI accumulation in mice that have hemochromatosis conditions. In Figure 2A, they show that Hfe−/− (type 1 hemochromatosis mice) that had ZIP14 functioning at 4 weeks of age have significantly elevated levels of non-heme iron in the liver, indicating that they are indeed storing excess iron there. However, these increased iron levels were not seen in the pancreas, heart, kidney, or spleen. On the other hand, the double-knockout mice (Hfe−/−; Slc39a14−/−) did not show this iron accumulation in the liver but instead loaded iron in the kidney. Moving on to Figure 2B, the researchers demonstrate that the lack of hepatic iron accumulation in Hfe−/−; Slc39a14−/− mice was not due to impaired dietary iron absorption. All groups, including the double-knockout mice, showed no significant differences in levels of iron absorption (Figure 2B), suggesting that the issue lies in the liver’s ability to store the absorbed iron (Figure 2C). Specifically, Figure 2C demonstrates a significant main effect of hemochromatosis on iron absorption, indicating that individuals with type 1 hemochromatosis (Hfe−/−) exhibit significantly higher iron absorption compared to those without the condition. Furthermore, the liver of the double knockout mice did not accumulate iron in it -although iron is absorbed- which indicates that the amount of absorbed iron is stored somewhere else rather than the liver. In addition, they used both Perls’ stain and DAB-enhanced Perls’ stain on the liver tissue from the different mice models to visually analyze the accumulation of iron (Figure 2 D-F). Perls’ stain detects ferric iron (Fe³⁺) by forming a blue pigment called Prussian blue. The DAB-enhanced version is more sensitive, using an enzyme (often peroxidase) to react with iron and produce a brown precipitate where iron is present. After staining the tissues’ sections, they found that only Hfe−/− mice expressed excessive iron accumulation, which is expected because these mice have hemochromatosis. Interestingly, once ZIP14 was knocked out in the hemochromatosis mice, there was a negligible amount of iron accumulation in the liver (Figures 2E–F). To further explore whether this lack of hepatic iron accumulation was related to plasma iron levels, they measured several parameters in Figures 2G to 2J, including plasma iron concentrations, total iron-binding capacity (TIBC), transferrin saturation, and NTBI. They found that plasma iron indices were not significantly affected by the loss of SLC39A14, indicating that the reduced liver iron accumulation in Hfe−/−; Slc39a14−/− mice was not due to lower plasma iron levels. Additionally, they measured the body weight of the mice, and the differences were not significant (Figure 2K).
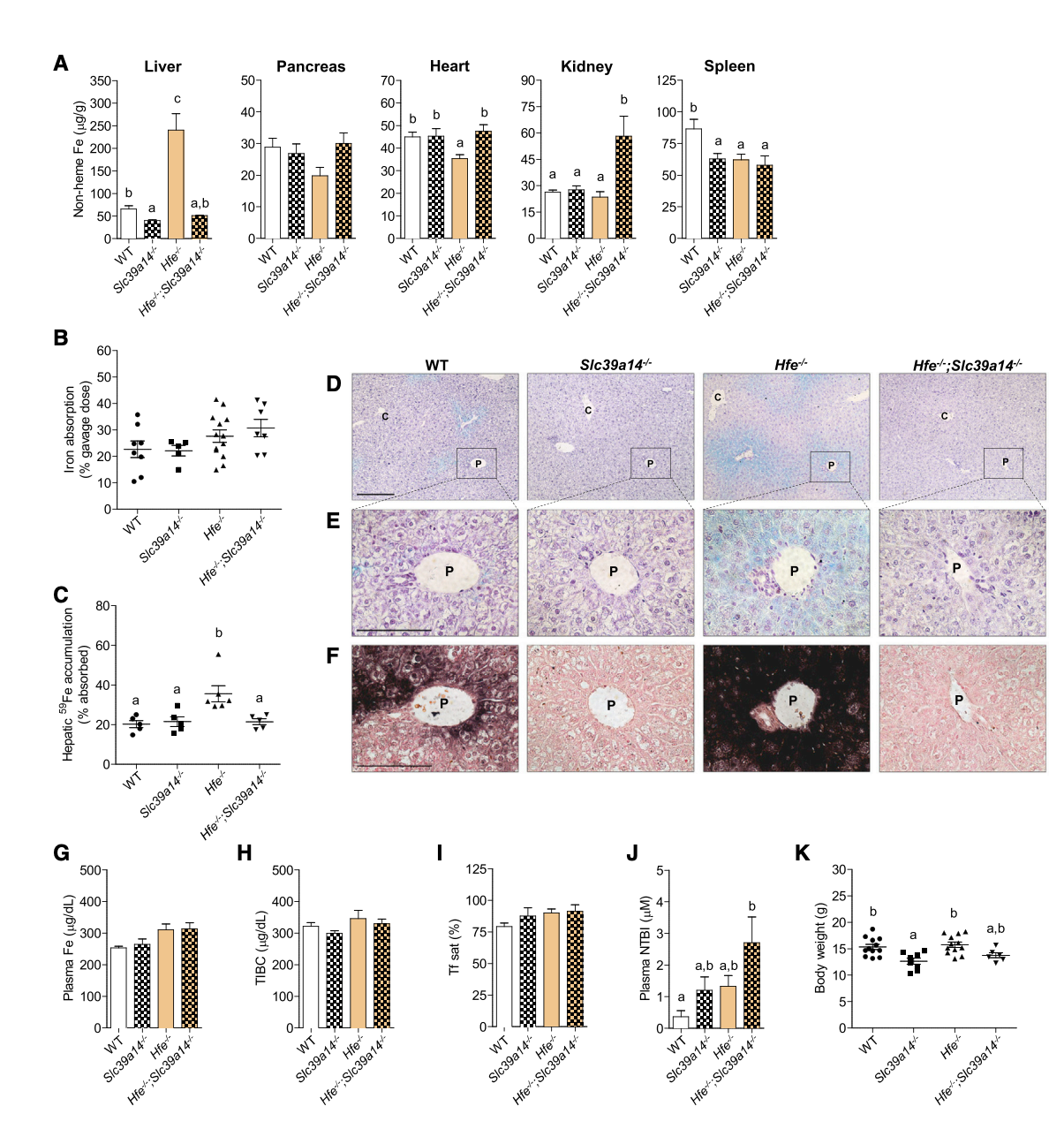
Figure 2
To examine the impact of losing SLC39A14 in severe hemochromatosis, researchers studied double-knockout Hfe2−/−; Slc39a14−/− mice, a model for juvenile (type 2) hemochromatosis. At 6 weeks, Hfe2−/− mice with functional ZIP14 showed a dramatic increase in liver and pancreas iron, and a 1-fold increase in heart iron compared to wild-type controls (Figure 3A). In double-knockout mice, liver and pancreatic iron decreased, and heart iron increased by 26%. Kidney and spleen iron levels rose significantly—2.6-fold and 11-fold, respectively—indicating ZIP14-independent iron accumulation (Figure 3A). Moreover, Iron absorption was elevated in Hfe2−/− mice and decreased by 25% in the double-knockout mice (Figure 3B). Hepatic iron accumulation was reduced in double-knockout mice, consistent with findings from Hfe−/−; Slc39a14−/− models (Figure 3C). Although pancreatic iron was higher in double-knockout mice compared to ZIP14-only knockouts, the result was not statistically significant due to the small sample size (n = 4). Perls’ and DAB-enhanced staining showed extensive iron deposits in hepatocytes of Hfe2−/− mice, while double-knockout mice had iron in nonparenchymal liver cells (Figure 3. F-H). Staining with the macrophage (white blood cells) marker F4/80 and Perls’ stain (Figure 4) revealed that some iron was stored in Kupffer cells (liver macrophages), suggesting that when ZIP14 is lost in type 2 hemochromatosis, iron is redirected to and stored in kupffer cells, which naturally store iron as part of their activity. Additionally, it was shown that the body weight of the mice with the double knockout decreased compared to the only hemochromatosis mice which might indicate that the overall health deterioration of type2- hemochromatosis mice due to the impairment of ZIP14. Additionally, loss of ZIP14 in the single- or double-knockout mice did not significantly affect total iron-binding capacity (TIBC), transferrin saturation, or plasma non-transferrin-bound iron (NTBI) concentrations, as displayed in Figures 3J–3L. Interestingly, mice lacking HFE2 exhibited higher plasma NTBI levels compared to their controls, and this is probably due to the impairment of the absorption paired with the high iron levels (Figure 3L).
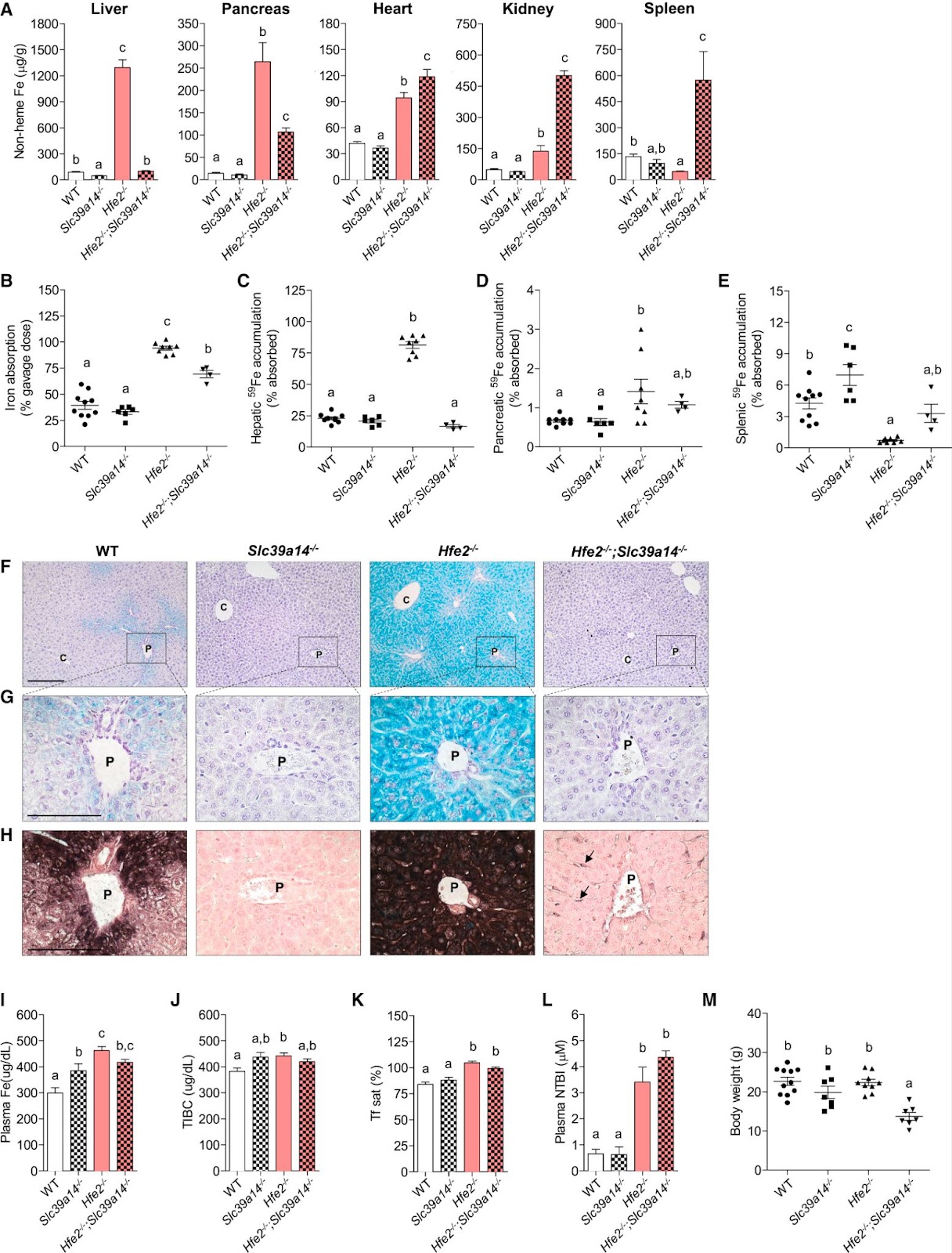
Figure 3
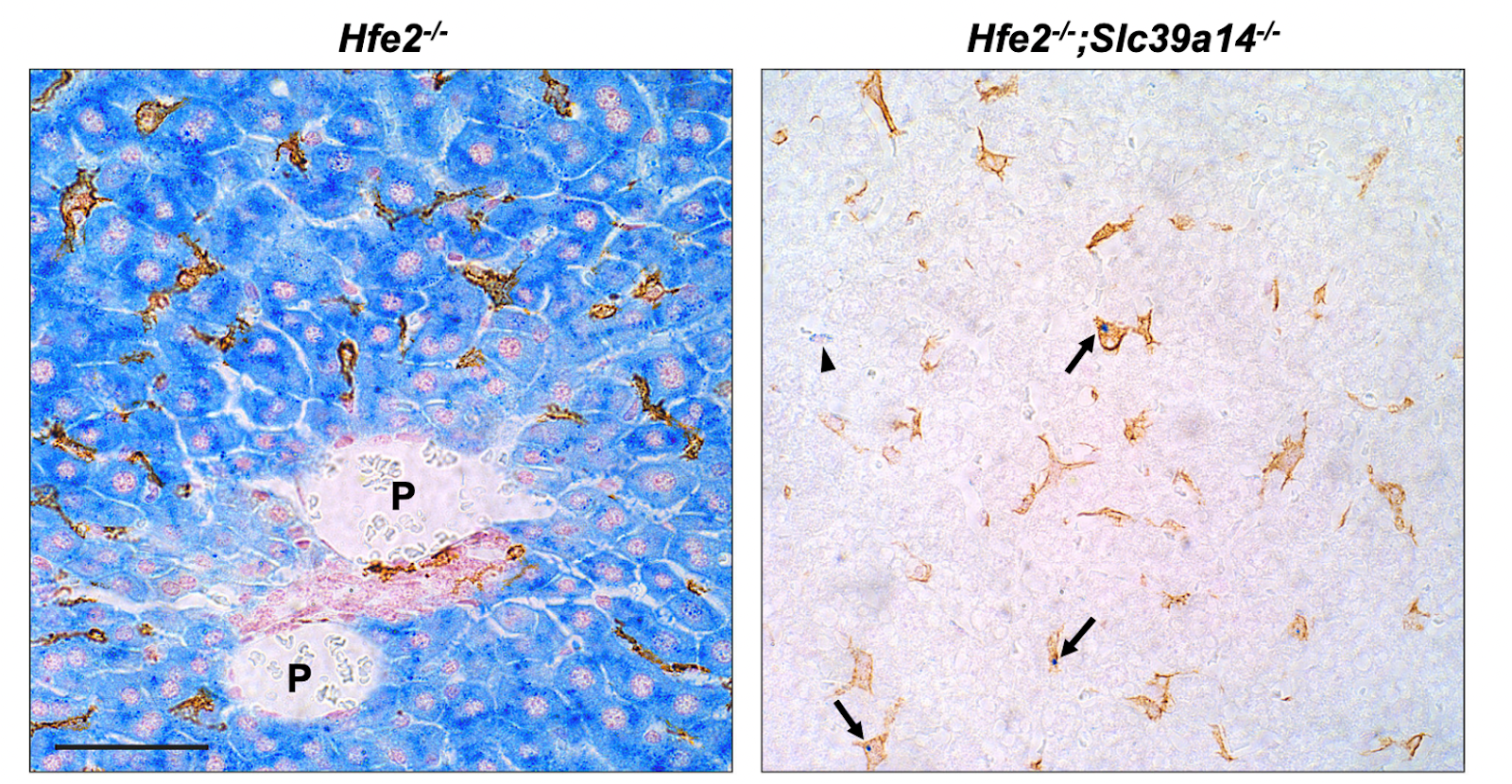
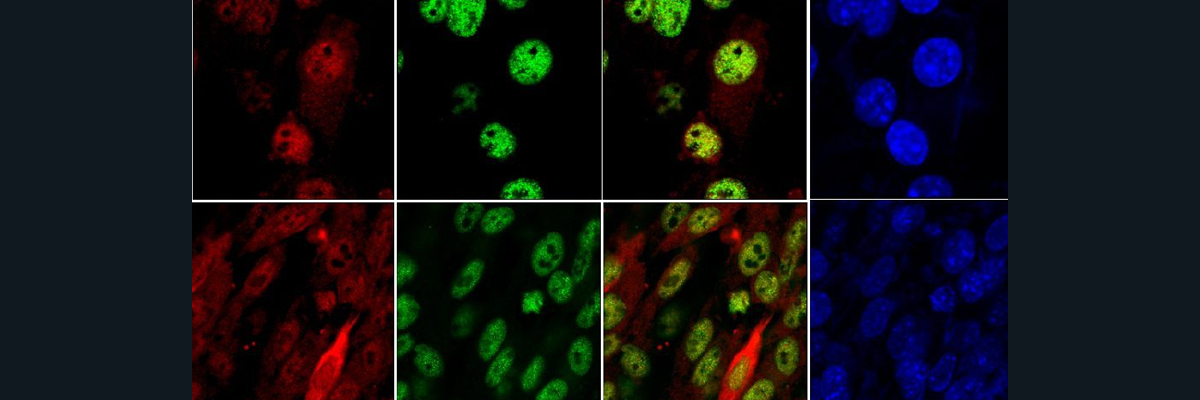
Figure 4. Staining the iron in the liver with perl’s staining (blue) and F4/80 (golden) for kupffer cells
In conclusion, this study highlights the critical role of ZIP14 in regulating iron accumulation in hemochromatosis. While these findings provide insight into potential therapeutic strategies targeting ZIP14 to reduce liver iron overload, such treatments are not yet feasible. Currently, individuals with hemochromatosis manage symptoms using medications such as iron chelators, which remove excess iron from the body. If patients are not anemic, phlebotomy (blood withdrawal) is another common treatment.
Looking ahead, genetic treatments for hemochromatosis could offer a cure if tools and therapies are developed and approved. Ongoing research shows promising progress in finding genetic solutions for this disease. A recent study conducted by Dr. Alice Rova in Dr. Michael Ott’s lab at Hannover Medical School demonstrated an effective genetic treatment for hemochromatosis in mice. Specifically, this study focused on correcting the C282Y mutation, which disrupts the HFE protein’s folding and function [9]. Researchers used the adenine base editing system (ABE7.10) (a gene editing tool) to convert adenine (A), the mutation-causing base, to guanine (G), the normal base. First, to validate their editing technique, they performed the HFE-GFP “Switch-on” system experiment. In this system, successful adenine-to-guanine editing results in GFP expression and cells’ glowing, while unedited mutations make the cell not glow. This experiment confirmed the effectiveness of ABE7.10 [8].
For the main experiment, they used two mouse models: healthy mice (WT) and those with the C282Y mutation (hemochromatosis mice). They delivered the correction system (ABE7.10) using a recombinant adeno-associated virus (AAV8) split-vector system. AAVs are commonly used in gene therapy due to their ability to efficiently deliver genetic material with relatively low immune responses [8]. After injecting the mice with the the vector, liver DNA from eight animals was analyzed at day 112 using Next-Generation Sequencing (NGS), showing a base conversion frequency (A to G) of 6.5% ± 2.3. After 112 days, serum analysis revealed significantly lower transferrin saturation (TS), indicating reduced circulating iron, and higher unbound iron binding capacity (UIBC), reflecting increased available transferrin for iron transport, in treated animals compared to controls (saline-injected mice). Hepcidin levels were significantly higher in treated hemochromatosis mice, indicating restored iron regulation. Liver tissue analysis showed no abnormalities in morphology using hematoxylin and eosin (H&E) staining. Prussian Blue staining revealed reduced iron accumulation in hepatocytes of treated animals compared to untreated hemochromatosis controls (Figure.5).
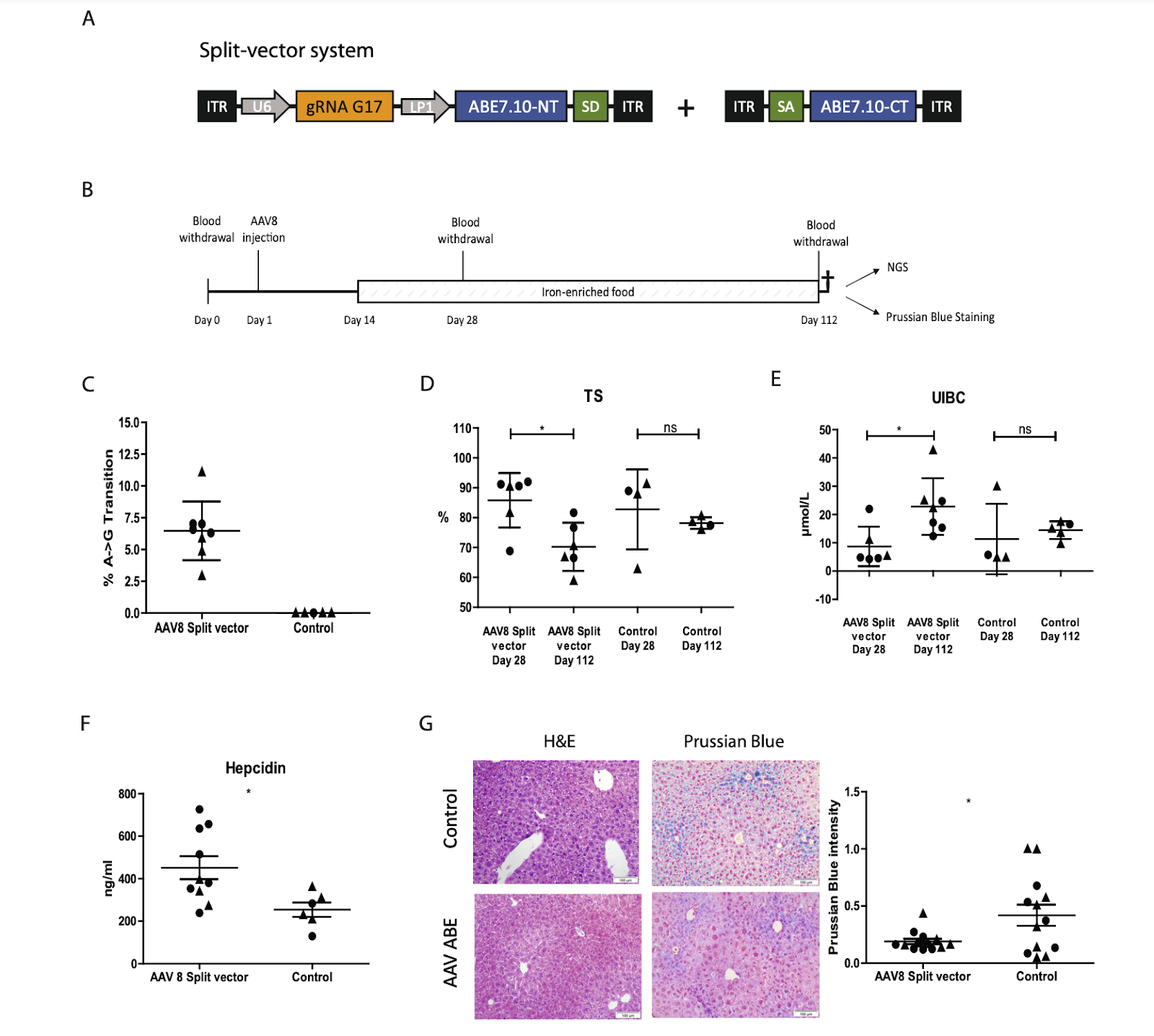
Figure.5
Upon injecting the mice with a higher-dose of split-vector containing the correction tool, researchers observed that after four months, iron accumulation in the hemochromatosis mice decreased significantly more than in the previous condition with a lower-dose split-vector injection, and this can be noted using the Prussian Blue staining (Figure.6 ).
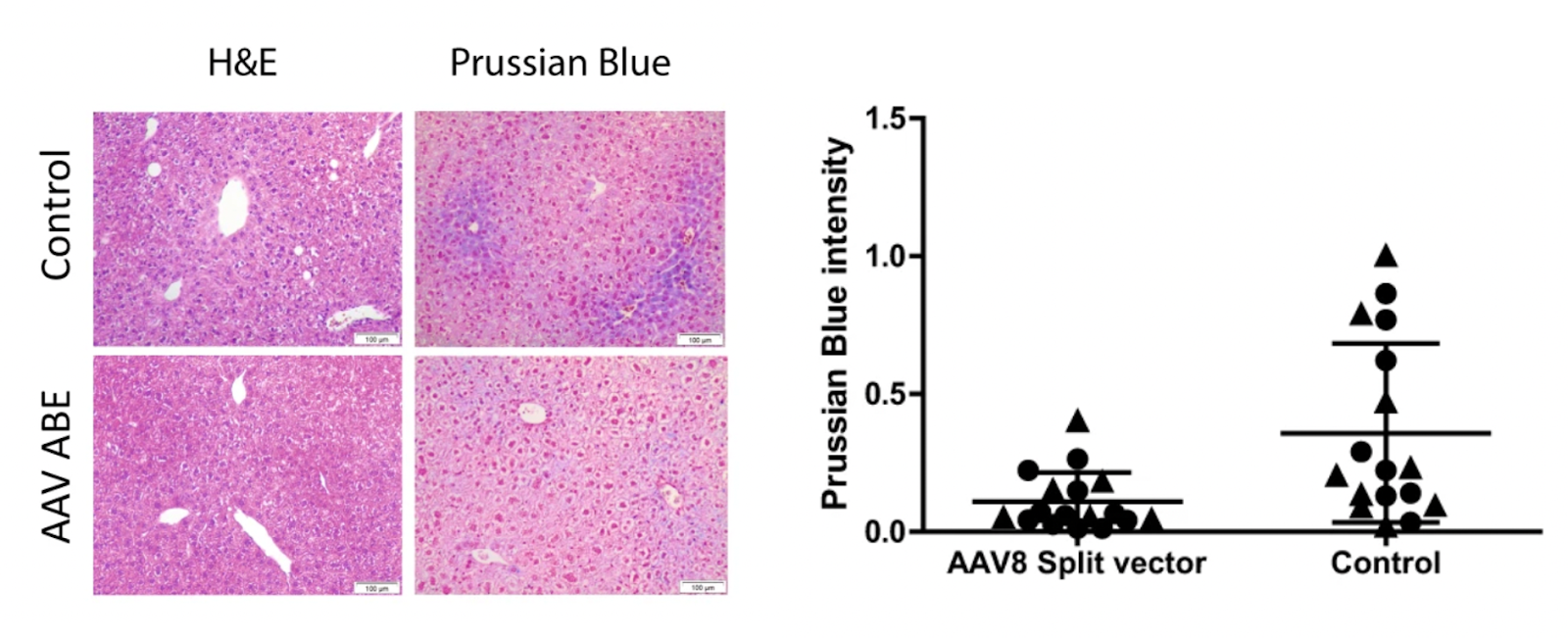
Figure.6
This study represents a promising step toward genetically treating type 1 hemochromatosis. However, further research, testing, and regulatory approvals are required before such therapies can be applied to humans. If successful in clinical trials, these therapies could eventually become a viable treatment option.
In summary, hemochromatosis, with all its types, primarily mainly targets the liver and results from disruptions in iron transport and regulatory proteins such as ZIP14 and HFE. Current treatments, such as iron chelation and blood withdrawal, are not sustainable long-term solutions. Without treatment, the disease can lead to fatal organ failure due to iron accumulation. Further research is essential to understand iron regulation and how its disruption leads to hemochromatosis. Studies like those by Jenkitkasemwong et al. provide critical insights into the disease’s mechanisms, while Rova et al. highlight potential genetic cures. Continued advancements in this field are vital to developing effective and lasting treatments for this challenging condition.
References:
[1] Bogdan, Alexander R., Masaki Miyazawa, Kazunori Hashimoto, and Yoshiaki Tsuji. 2016. “Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease.” Trends in Biochemical Sciences 41 (3): 274–86. https://doi.org/10.1016/j.tibs.2015.11.012.
[2] Hooda, Jagmohan, Ajit Shah, and Li Zhang. 2014. “Heme, an Essential Nutrient from Dietary Proteins, Critically Impacts Diverse Physiological and Pathological Processes.” Nutrients 6 (3): 1080–1102. https://doi.org/10.3390/nu6031080.
[3] Wallace, Daniel F. 2016. “The Regulation of Iron Absorption and Homeostasis.” The Clinical Biochemist. Reviews 37 (2): 51–62.
[4] Drakesmith, Hal, Emma Sweetland, Lisa Schimanski, Jon Edwards, Diana Cowley, Mubeen Ashraf, Judy Bastin, and Alain R. M. Townsend. 2002. “The Hemochromatosis Protein HFE Inhibits Iron Export from Macrophages.” Proceedings of the National Academy of Sciences 99 (24): 15602–7. https://doi.org/10.1073/pnas.242614699.
[5]“Canadian Liver Foundation.” 2017. Canadian Liver Foundation. 2017. https://www.liver.ca/patients-caregivers/liver-diseases/hemochromatosis/.
[6] Brissot, Pierre, Antonello Pietrangelo, Paul C. Adams, Barbara de Graaff, Christine E. McLaren, and Olivier Loréal. 2018. “Haemochromatosis.” Nature Reviews Disease Primers 4 (4): 18016. https://doi.org/10.1038/nrdp.2018.16.
[7]Nguyen-Lefebvre, Anh Thu, and Anatolij Horuzsko. 2015. “Kupffer Cell Metabolism and Function.” Journal of Enzymology and Metabolism 1 (1): 101. https://pmc.ncbi.nlm.nih.gov/articles/PMC4771376/.
[8]Zhao, Longmao, Zixuan Yang, Minghui Zheng, Lei Shi, Mengyun Gu, Gang Liu, Feng Mao, Cheng Yan, Fengtao Huang, and Naping Tang. 2024. “Recombinant Adeno-Associated Virus 8 Vector in Gene Therapy: Opportunities and Challenges.” Genes and Diseases 11 (1): 283–93. https://doi.org/10.1016/j.gendis.2023.02.010.