Friedreich Ataxia (FRDA) and Aceruloplasminemia are both autosomal recessive disorders with severe impacts on the human body. They underscore the importance of metal homeostasis in the body, more specifically how crucial metals like iron and copper, although essential for numerous biological processes, can cause severe harm when misregulated.
Friedreich Ataxia results from a genetic mutation affecting frataxin, a mitochondrial protein involved in iron regulation. These mutants stem from iron accumulation in mitochondria, which causes oxidative stress and damage to cells, usually affecting the nervous system and heart. The first research paper investigates how frataxin deficiency affects adipose tissue function and overall metabolic health. Researchers utilized the YG8R mouse model, which mimics the genetic and phenotypic characteristics of FRDA. The findings revealed that a reduction of FXN expression disrupted lipid metabolic homeostasis, a primary cause of insulin resistance in individuals with FRDA.
The study highlighted a critical link between FXN expression and lipolysis (a part of the lipid homeostasis pathway) in adipose tissue. FXN deficiency was associated with impaired lipolysis, which is an essential process for mobilizing energy stored in fat. This impairment could contribute to insulin resistance, a common complication in FRDA patients. The research also indicated that FXN deficiency decreased energy production and increased oxidative stress due to iron accumulation. This oxidative stress is detrimental to cellular health and contributes to the neurodegenerative aspects of FRDA.
Overall, the paper emphasizes FXN’s role in maintaining metabolic health by regulating iron metabolism and mitochondrial function. The interconnectedness between FXN expression, lipolysis, and insulin sensitivity is crucial for understanding the metabolic complications associated with FRDA. These insights pave the way for potential therapeutic strategies targeting iron dysregulation and oxidative stress in FRDA patients. Findings such as these highlight the importance of further research into the mechanisms of iron metabolism in FRDA, as investigating potential treatments, such as iron chelation therapy and antioxidants, could offer new avenues for managing this debilitating disease.

This figure demonstrates how the researchers determined a remodeling of the entire lipid metabolism pathway. Lipolysis and FA beta-oxidation were suppressed, and lipogenesis was enhanced due to Frataxin (FXN) deficiency.

In this experiment, they realized that a deficiency of FXN causes an increase in inflammatory factors and cell oxidative stress in the white and brown adipose tissues of the mutated mice.

The researchers noted that FXN deficiency aggravated obesity (induced by a high-fat diet), diabetes, and liver steatosis (increased fat build-up) at a young age in the mouse model.
Aceruloplasminemia involves a deficiency in ceruloplasmin, a copper-binding protein crucial for iron mobilization. In the absence or reduction of ceruloplasmin, iron accumulates in various organs, including the brain, liver, and pancreas, leading to neurological impairment, diabetes, and liver disease. In a recent study, researchers aimed to analyze the metabolic issues associated with aceruloplasminemia using a ceruloplasmin-deficient mouse model (CpKO). The CpKO mice were treated with purified ceruloplasmin to evaluate the potential therapeutic effects of ceruloplasmin replacement.
The paper noted that the CpKO mice exhibited significant weight gain and increased adipose tissue accumulation compared to WT mice, indicating a clear disruption in lipid metabolism. Furthermore, the researchers observed increased macrophage infiltration (a type of white blood cell) in the adipose tissue and liver of CpKO mice, linking chronic inflammation to metabolic dysregulation. The ceruloplasmin injection treatment reduced iron accumulation in the liver and steatosis (a condition where excess fat builds up in the liver). However, the treatment did not fully normalize the expression of iron homeostasis-related proteins. The ceruloplasmin replacement therapy also limited macrophage infiltration, reduced serum triglyceride levels (indicative of a functioning lipid metabolism system), and partially restored levels of key cell-signaling molecules in the adipose tissue.
Overall, the study highlights ceruloplasmin’s crucial role in maintaining metabolic balance and its involvement in the dysregulation of lipid metabolism in aceruloplasminemia. The findings suggest that ceruloplasmin replacement therapy may offer a promising strategy to limit the systemic effects of this disorder. This research allows us to learn more about this under-studied disease and opens the door for future studies that aid in developing effective treatments for this challenging condition.

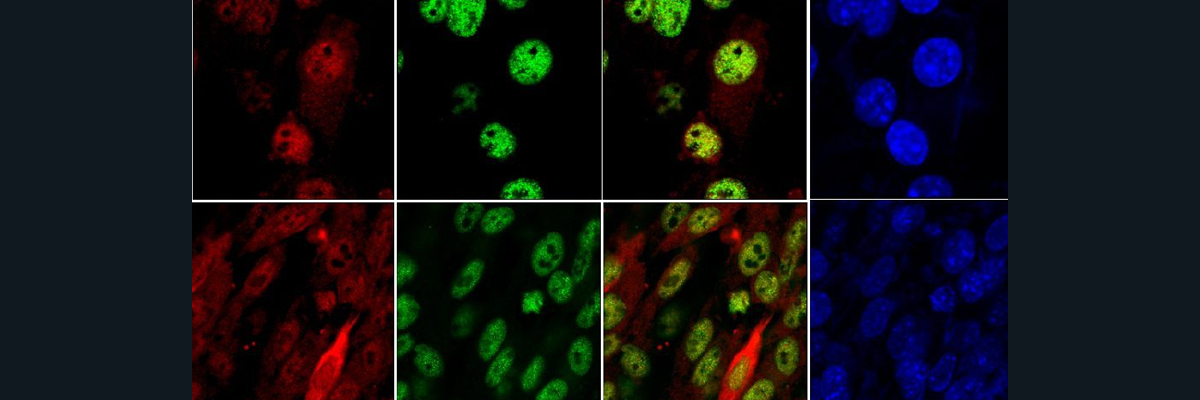
This figure shows that the ceruloplasmin deficient (CpKO) mice exhibit liver steatosis and macrophage infiltration. However, an administration of ceruloplasmin via injection decreases these symptoms.

In this experiment, the researchers found that the CpKO mice show altered adipokines, which are cell-signaling molecules produced by the adipose tissue that play functional roles in the energy/metabolic status of the body, inflammation, and obesity. These altered states are partially restored following ceruloplasmin administration. Thus, Cp administration can be further studied as a potential treatment for aceruloplasminemia.
Sources:
Wu, L., Huang, F., Yang, L., Yang, L., Sun, Z., Zhang, J., Xia, S., Zhao, H., Ding, Y., Bian, D., & Li, K. (2024). Interplay of FXN expression and lipolysis in white adipocytes plays a critical role in insulin sensitivity in Friedreich’s ataxia mouse model. Scientific Reports, 14(1), 1-14. https://doi.org/10.1038/s41598-024-71099-7
Raia, S., Conti, A., Zanardi, A., Ferrini, B., Scotti, G. M., Gilberti, E., De Palma, G., David, S., & Alessio, M. (2023). Ceruloplasmin-Deficient Mice Show Dysregulation of Lipid Metabolism in Liver and Adipose Tissue Reduced by a Protein Replacement. International journal of molecular sciences, 24(2), 1150. https://doi.org/10.3390/ijms24021150