Acrodermatitis enteropathica (AE) is a disorder that results from zinc (Zn) deficiency in the cell. AE results from mutations in the SLC39A4 gene, which encode the Zip4 protein, a protein that brings Zn into the cytosol. It is most prevalent in babies, particularly when they are being weaned off breast milk. A second cause of AE is when the mother has a mutation in the SLC30A2 gene, which encodes ZnT2, which helps export Zn and attach it to carriers, specifically the mother is deficient in this gene in breast tissue, however, this version of the condition is not explored in this paper. Additionally, one can acquire Zn deficiency at any time. The prevalence of the condition is 1-9:1,000,000, making it extremely rare. The typical treatment for the disorder is Zn supplementation, which usually yields a full recovery.
The mutations that cause AE that are explored here are damaging to the way that Zip4 folds and locates itself. If a protein folds incorrectly, it could lose its function, which occurs here. Improper folding and mutations cause the complete loss of importing Zn into the cell. Moreover, these mutations in the region of the protein that exist in the extracellular domain cause it to localize to the wrong area of the cell (in part, result of the signal that should tell the protein to go where it needs to being prematurely done in these mutations). Instead of going to the plasma membrane, the protein remains inside of the cell, meaning that it cannot engage in its intended function of moving Zn into the cell, since it is not on the plasma membrane (Fig1. A,B).
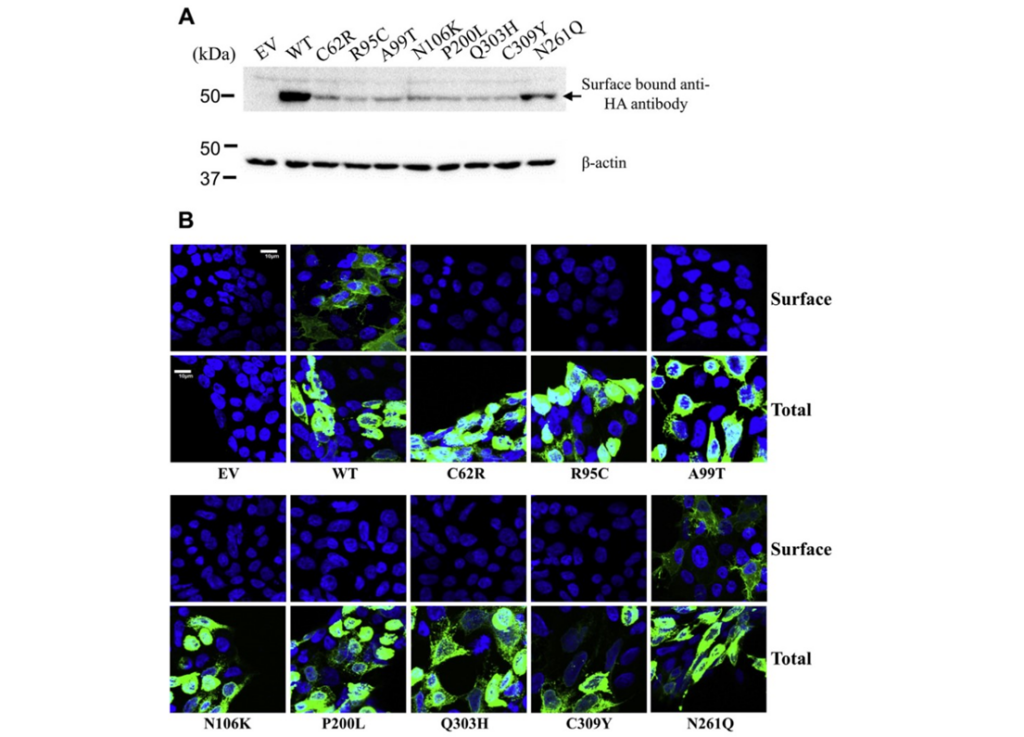
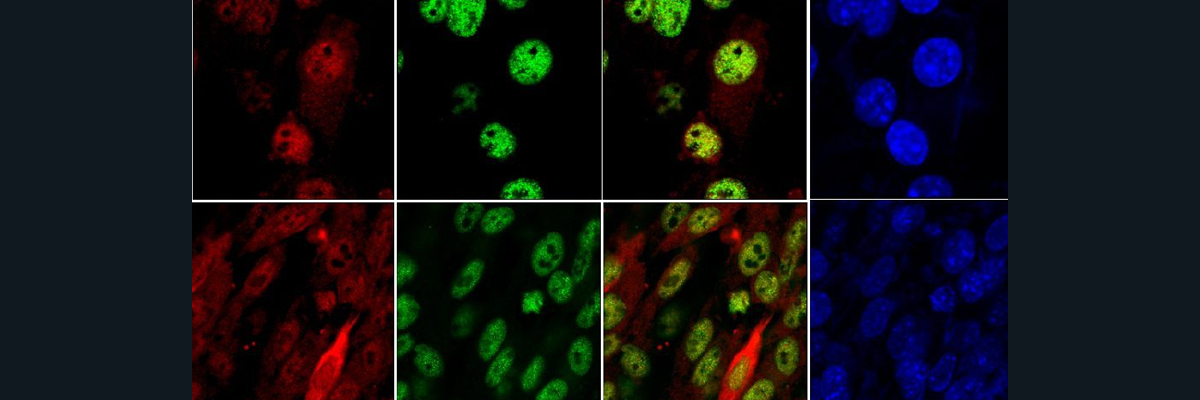
Figure 1: A shows the amount of protein that exists on the cell surface between the healthy patients and the AE mutations. B depicts imagery of the localization of Zip4 protein, where in the healthy patient, Zip4 localizes to the plasma membrane in a greater quantity than the mutants, which show most Zip4 is located within the cell.
Moreover, they analyzed the structure of the protein’s mutants and determined that every one had very different secondary structures from the protein’s manifestation in healthy patients (Fig2. A). Additionally, the mutants lost their structure when exposed to heat at a lower temperature than their wild type counterpart, further indicating the instability of the mutant proteins (Fig2. C).
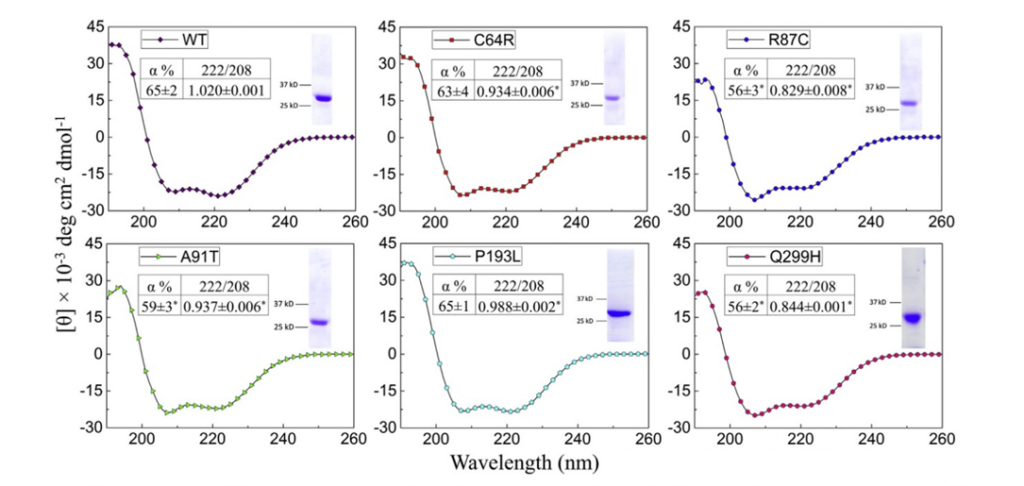
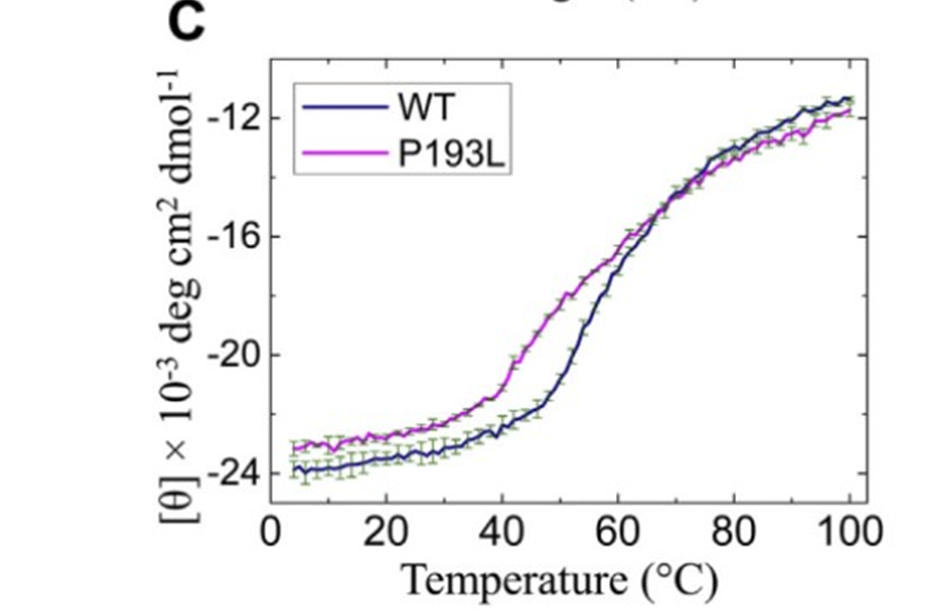
Figure 2: A shows the changes in the secondary structure of the wild type versus the mutant. C demonstrates the greater stability of the wild type versus the mutants, particularly versus the P193L mutant.
Therefore, there are a few main conclusions of this paper. First, the signal that indicates where the protein should go being placed earlier in the protein than it should causes the protein to exist inside of the cell, instead of the plasma membrane. Second, these mutations cause structural, functional, localizable, and expression problems for the protein. In the future, it may be wise to explore how helping this protein fold properly and localize correctly could help cure those with AE.
Citations:
Jagadeesan, Soumya, and Feroze Kaliyadan. “Acrodermatitis Enteropathica.” National Institutes of Health, U.S. National Library of Medicine, 3 Apr. 2023, www.ncbi.nlm.nih.gov/books/NBK441835/. Accessed 26 Sept. 2024.
Kuliyev, Eziz et al. “Zinc Transporter Mutations Linked to Acrodermatitis Enteropathica Disrupt Function and Cause Mistrafficking.” The Journal of Biological Chemistry, U.S. National Library of Medicine, Jan. 2021, pubmed.ncbi.nlm.nih.gov/33837739/. Accessed 26 Sept. 2024.