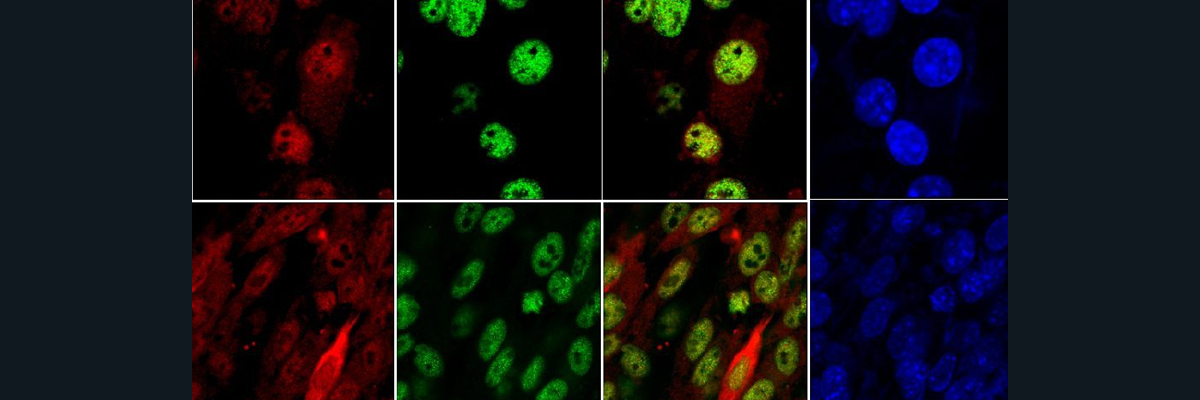
Amyotrophic Lateral Sclerosis, or ALS, is a neurodegenerative disease primarily affecting the motor cortex and brain stem nuclei, leading to muscle atrophy, and sometimes language problems, behavioral changes and executive disfunction. Cytoskeletal defects leading to impaired neuronal transport, changes in RNA metabolism resulting in protein aggregates, impaired autophagy and mitochondrial dysfunction all serve as cellular mechanisms through which ALS may develop, contributing to the degeneration of neurons. These cellular mechanisms may occur through a variety of mutations, one of which affects the SOD1 protein and results in its aggregation.
Typically, SOD1 contains copper and zinc cofactors. The copper cofactor has catalytic function, playing an active role in converting superoxide to hydrogen peroxide and oxygen – one of the chief roles of SOD1. The zinc cofactor of SOD1 is believed to play a primarily structural role in ensuring the integrity of SOD1. In order to examine the role of these metal cofactors in maintaining the structure of SOD1 and preventing SOD1 aggregation.
To observe SOD1 aggregation in the presence and absence of copper and zinc, H72F and H121F mutations were induced to prevent zinc and copper binding, respectively. These mutants were examined alongside wild type and apo-SOD1 variants, with the apo-SOD1 mutant lacking both metal cofactors. Structural similarities between these variants were also assessed. Additionally, due to prior research surrounding the preference of SOD1 mutants to aggregate in the presence of a cellular membrane, researchers measured SOD1 variant aggregates in the presence of artificial vesicles.
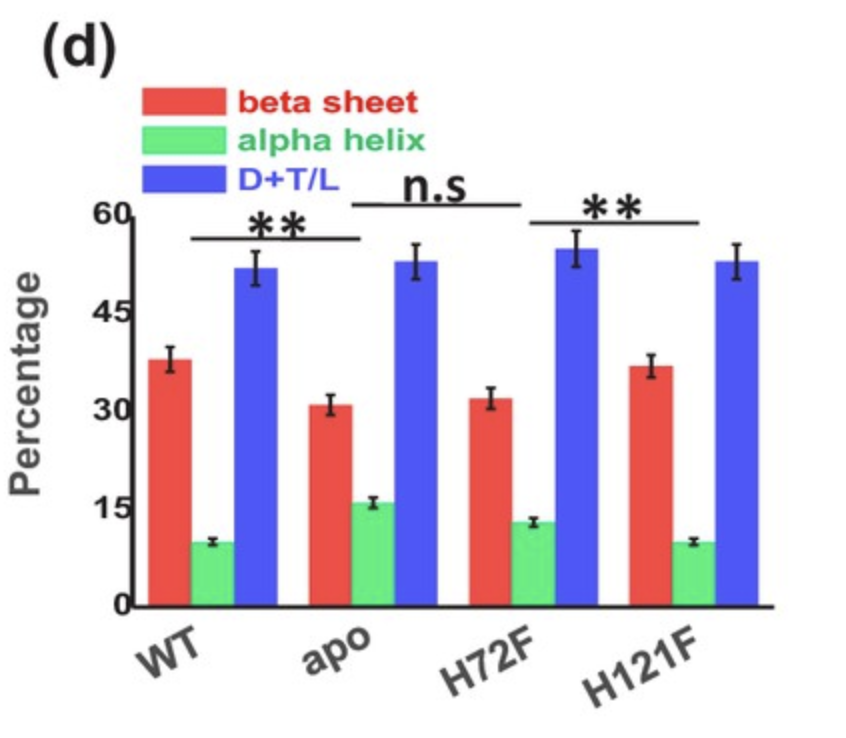
When assessing structural motifs, the content of alpha helices, beta sheets and turns and loops were measured as a percentage of the overall structure of the protein. Statistically insignificant differences in structural content were observed between apo-SOD1 and the H72F variant, as well as between wild-type SOD1 and the H121F variant (Figure 1). These results suggest the crucial role of zinc in ensuring the correct folding of SOD1, while additionally suggesting a far more insignificant role of copper in SOD1 folding.
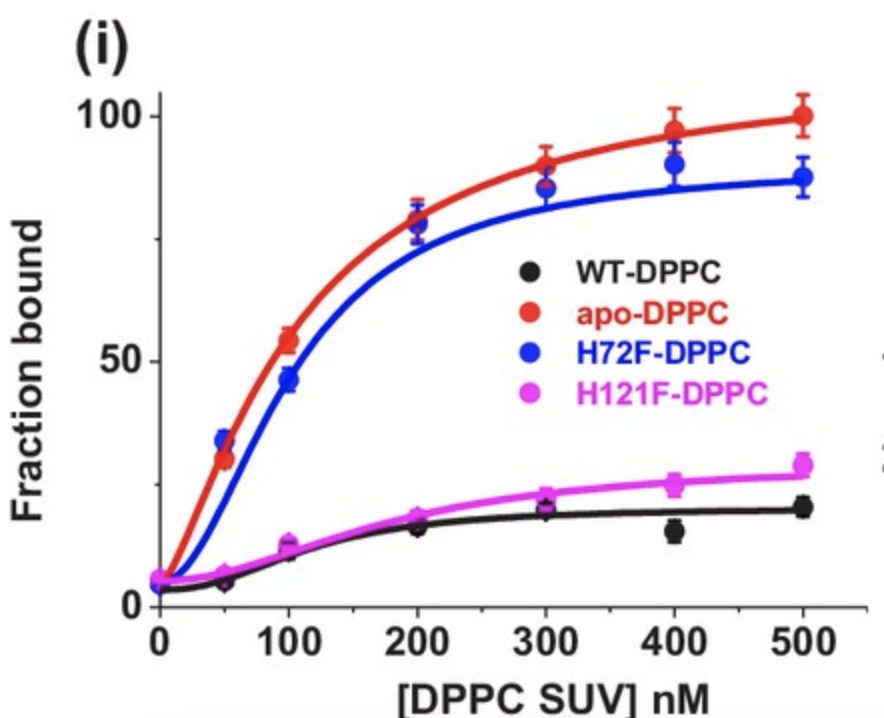
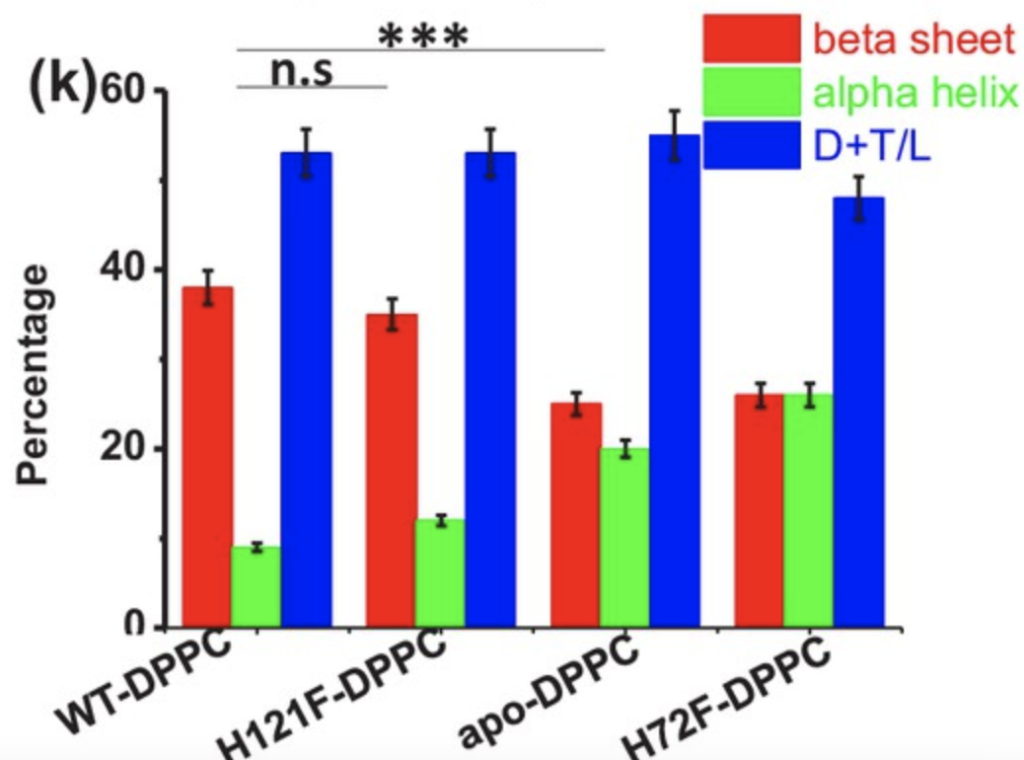
Binding curves were created for each of the four SOD1 variants, using DPPC SUVs to as artificial vesicles to simulate membrane binding. The apo-SOD1 and H72F variants bound the vesicles with far greater affinity than the wild type and H121F variant (Figure 2). Additionally, once bound to the membrane, structural similarities were measured, with similarities once again observed between apo-SOD1 and H72F, as well as H121F and the wild type (Figure 3). These findings point towards the lack of the zinc cofactor in the apo-SOD1 and H72F variants as a potential reason for the membrane binding ability observed in mutant SOD1, allowing aggregation.
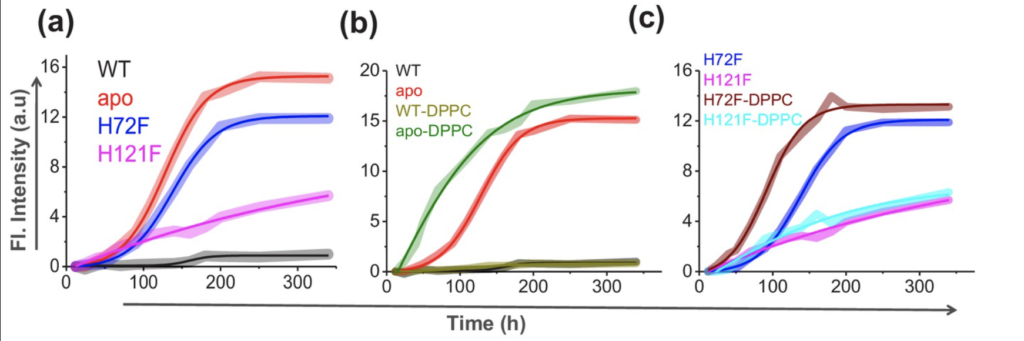
To directly measure SOD1 aggregation, thioflavin T fluorescence was utilized. Thioflavin T fluoresced upon binding protein aggregates, allowing visualization of the aggregation of each SOD1 variant. Fluorescence intensity for the apo-SOD1 and H72F samples dwarfed those of the wild type and H121F samples. Additionally, when bound to DPPC, simulating the membrane, apo-SOD1 and the H72F mutant exhibited greater fluorescence than samples lacking DPPC (Figure 4). This underlines the importance of zinc in preventing SOD1 misfolding and thereby preventing SOD1 aggregation, while also underscoring the importance of the cell membrane in encouraging SOD1 aggregation.
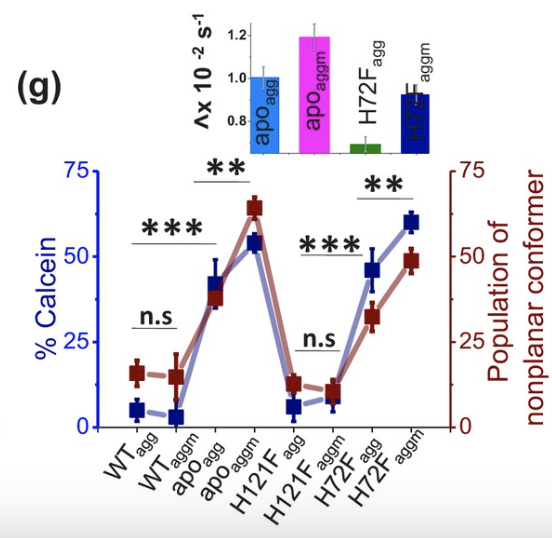
To measure the toxicity of the SOD1 aggregates, calcein was injected into vesicles and its leakage monitored, as aggregates may often induce membrane leakage. The fluorescence of calcein was measured to visualize leakage. Additionally, calcein adopts a nonplanar conformer upon exposure to aggregates, allowing a separate means of visualizing aggregate formation and toxicity of the samples. Each sample was measured with and without bound DPPC. The apo and H72F variants induced significantly more calcein leakage and nonplanar calcein conformers than the wild type and H121F variants, while DPPC-bound apo and H72F samples induced statistically greater calcein leakage and nonplanar conformers than their unbound counterparts (Figure 5). Toxicity of SOD1 aggregates and the ability of SOD1 to aggregate is significantly increased when SOD1 lacks its zinc cofactor, inducing misfolding conducive to aggregation. Additionally, this paper demonstrates the greater toxicity of membrane-bound SOD1 aggregates than those not bound to membranes.
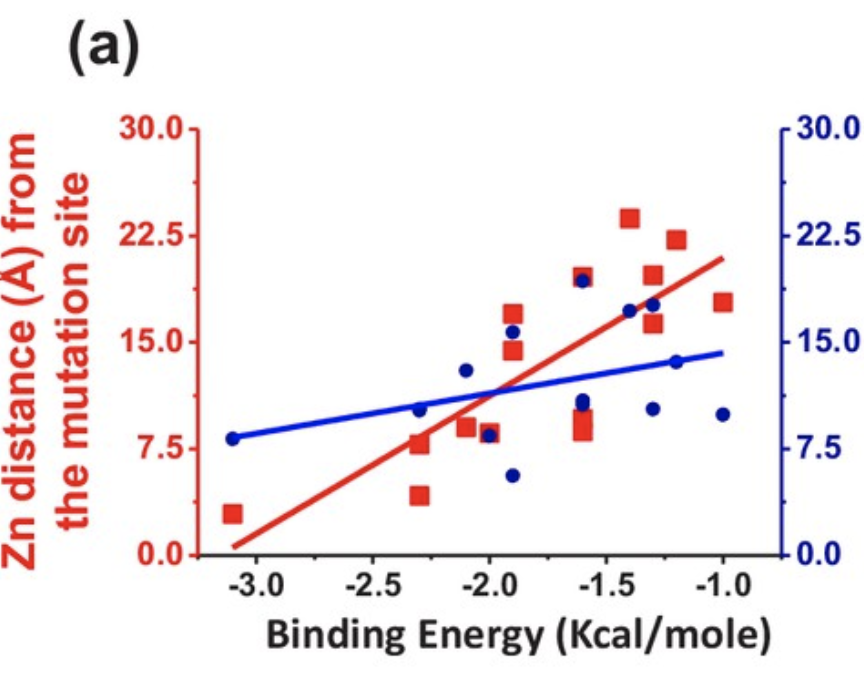
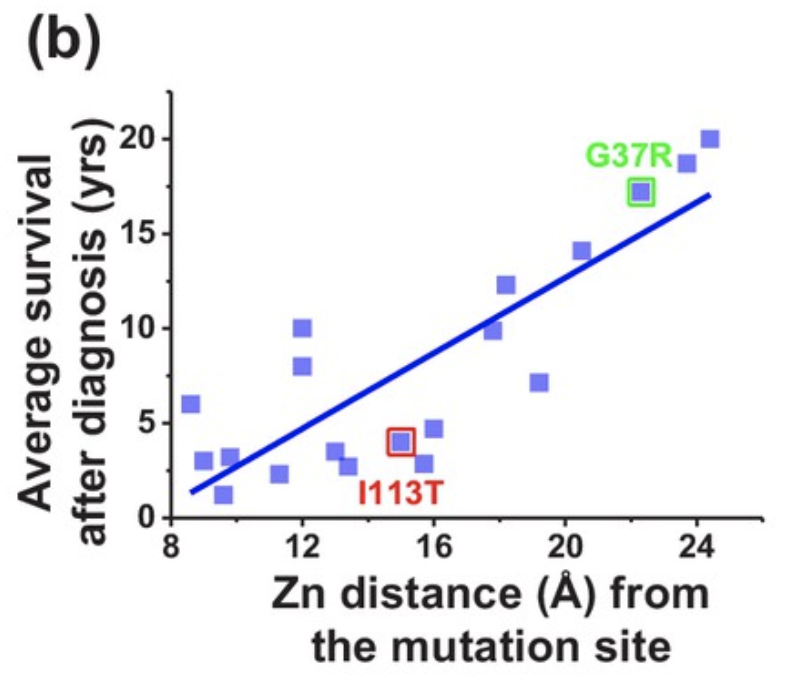
Finally, binding kinetics of zinc and copper were assessed based on the position of the mutation relative to the respective binding sites of the two metals. The binding energy between zinc and the protein decreased when the mutation occurred closer to the zinc binding site (Figure 6), effectively decreasing the chance of successful zinc binding. Additionally, mutations closer to the zinc binding site were associated with a lower average survival time following diagnosis (Figure 7), demonstrating the crucial role of zinc in preventing SOD1 aggregation leading to ALS.
SOD1 lacking its zinc cofactor, induced in this paper through a H72F mutation, resembles completely misfolded apo-SOD1 in its structure, ability to aggregate, ability to bind cellular membranes and toxicity upon aggregation, demonstrating the necessity of zinc in ensuring proper folding of SOD1. Additionally, access to the membrane increases SOD1 aggregation and toxicity. While a variety of mutations may result in ALS, ALS induced by SOD1 misfolding presents a potential drug target in SOD1 lacking its zinc cofactor.
Sources:
Sannigrahi, A., Chowdhury, S., Das, B., Banerjee, A., Halder, A., Kumar, A., Saleem, M., Naganathan, A. N., Karmakar, S., & Chattopadhyay, K. (2021). The metal cofactor zinc and interacting membranes modulate sod1 conformation-aggregation landscape in an in vitro ALS model. ELife, 10. https://doi.org/10.7554/elife.61453
Masrori, P., & Van Damme, P. (2020). Amyotrophic lateral sclerosis: a clinical review. European journal of neurology, 27(10), 1918–1929. https://doi.org/10.1111/ene.14393