Alzheimer’s Disease is a neurodegenerative disease most commonly characterized through the formation of beta-amyloid (Aβ) plaques and the development of neurofibrillary tangles caused by the hyperphosphorylation of the tau protein. Both of these pathways ultimately result in the formation of protein aggregates that lead to the degeneration and loss of neurons. Zinc plays a role in the formation of Aβ plaques and is additionally associated with the formation of hyperphosphorylated tau proteins, and typically mutations in zinc transporters designed to export zinc from neurons and inhibit release across the synapse are exhibited in Alzheimer’s Disease, contributing to the formation of extracellular protein aggregates. Investigation surrounding zinc concentration and NLRP3 activity, however, point towards a separate mechanism for the development of Alzheimer’s disease, independent from the canonical Aβ plaque formation.
To assess the effect of zinc concentration on Alzheimer’s Disease progression, researchers assigned mice to normal zinc (ZN), zinc-deficient (ZD) and zinc-deficient transitioned to normal zinc after three months (ZD/ZN). Wild-type and APP/PS1 mice were assessed, with the APP/PSI mutation designed to induce Alzheimer’s Disease. The mice performed Y-maze memory tasks and novel smell recognition tests to assess their cognitive abilities to measure disease progression, while brain tissue samples were additionally taken to assess the Aβ plaque burden. Finally, to assess the effect of NLRP3 activation on disease progression, NLRP3-containing mice (NLRP3 +/+) and NLRP3 knockout mice (-/-) were subjected to the experimentation conditions described above.
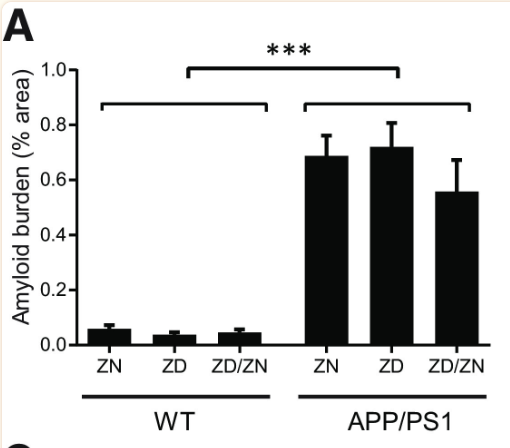
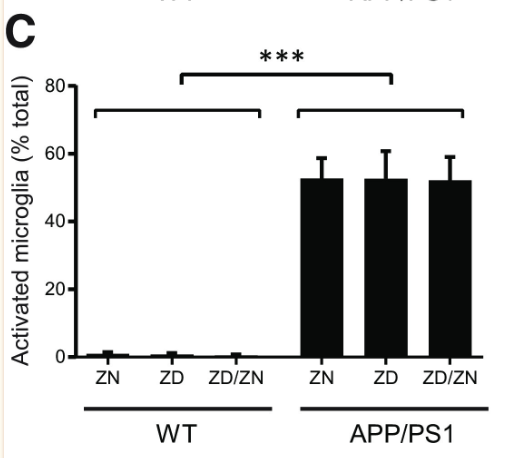
Zinc deficiency was found to have no statistically significant effect on Aβ plaque formation (Figures 1 and 2). While APP/PS1 mice did display a significantly higher Aβ plaque burden than the wild-type mice, zinc deficiency displayed no discernible role in promoting Aβ plaque formation (Figure 1). Additionally, no statistically significant effect was found in microglia activation between the ZN, ZD and ZD/ZN mice (Figure 2). Microglia activation typically occurs as an immune response to protein aggregation exhibited in Alzheimer’s disease, and serves as another way to visualize disease progression.
Despite the lack of correlation between zinc intake and Aβ plaque burden, ZD APP/PSI mice displayed statistically significant cognitive decline relative to ZN APP/PS1 mice (Figures 4 and 5). These results suggest a mechanism for Alzheimer’s Disease progression independent from Aβ plaque formation, accentuated by zinc deficiency.
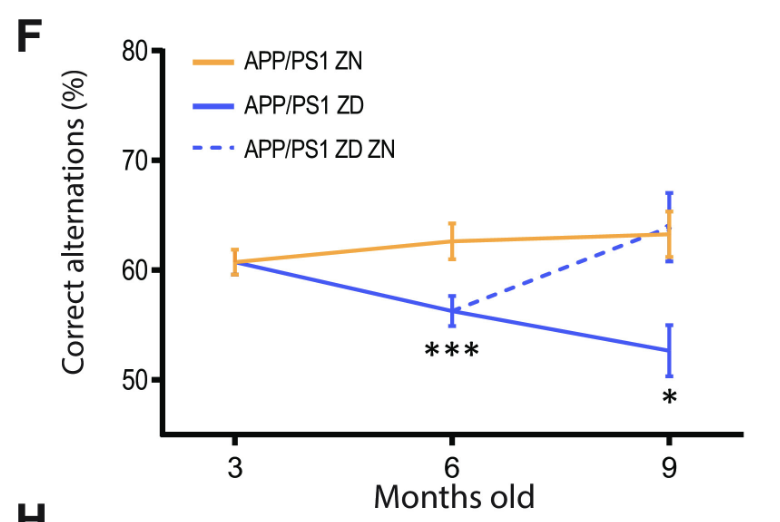
Figure 4: The percentage of correct alternations taken in the Y-maze test by APP/PS1 mice with varying zinc diets. A decrease in correct alternations represents cognitive decline. ZD mice exhibit a statistically significant cognitive decline relative to ZN mice, while ZD/ZN mice initially exhibit significant cognitive decline when on ZD diets, while exhibiting recovery once switched to ZN diets.
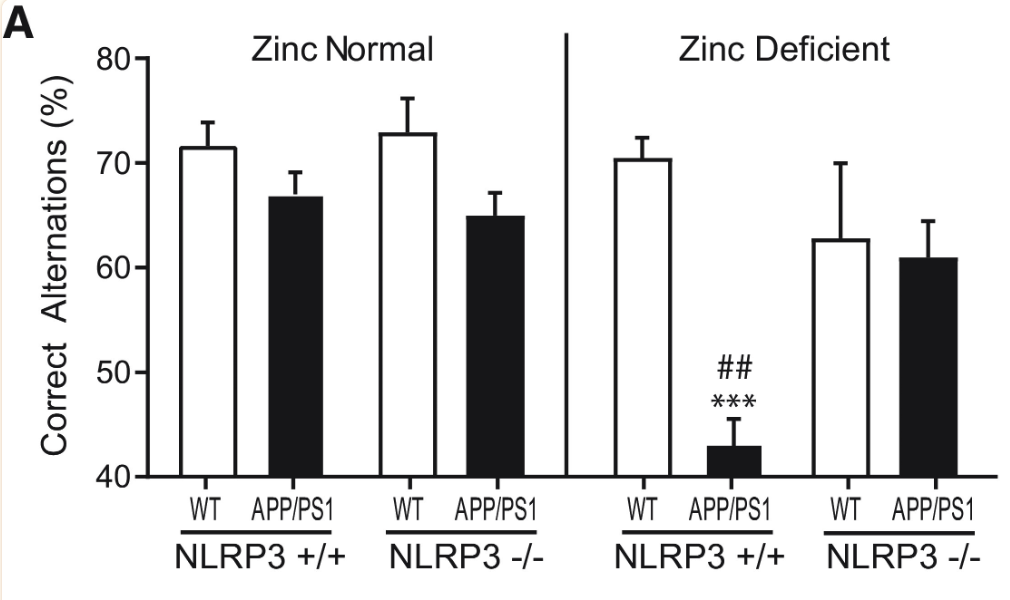
To assess the role of NLRP3 activation in Alzheimer’s Disease Progression, NLRP3 +/+ and NLRP3 -/- wild-type and APP/PS1 mice were treated with normal zinc and zinc-deficient diets. Notably, NLRP3 +/+, APP/PS1, zinc-deficient mice exhibited a far lesser percentage of correct alternations in the Y-maze test compared to other mice, indicating steep cognitive decline and linking NLRP3 activation to zinc deficiency and linking NLRP3 activation in Alzheimer’s Disease to cognitive decline (Figure 6).
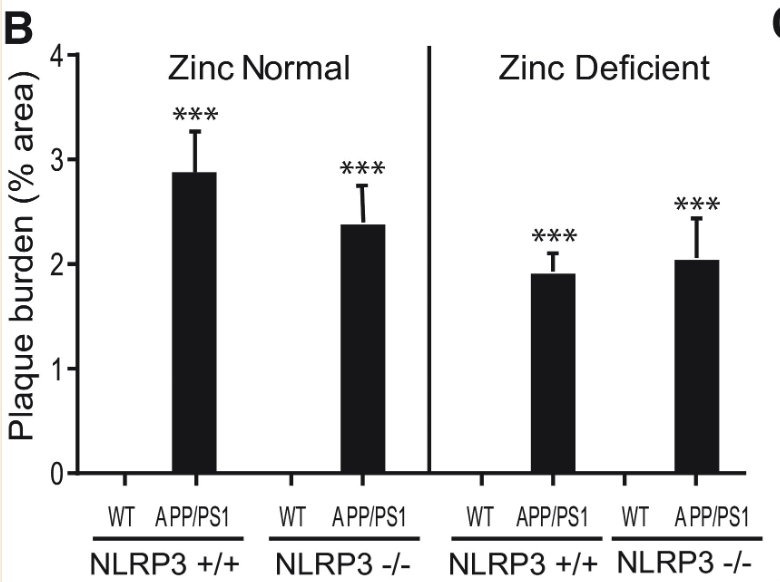
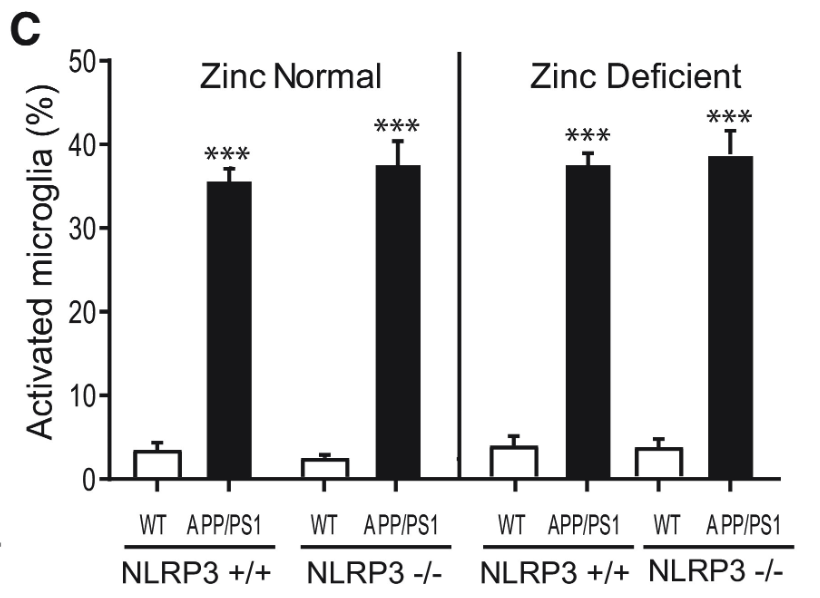
Plaque burden and microglia activation were measured once more with NLRP3 +/+ and NLRP3 -/- mice. While the plaque burden in APP/PS1 mice was significantly greater than in wild type mice, NLRP3 activity displayed no statistically significant correlation to plaque burden, further pointing towards NLRP3-dependent Alzheimer’s Disease progression being independent from Aβ plaque formation (Figures 7 and 8).
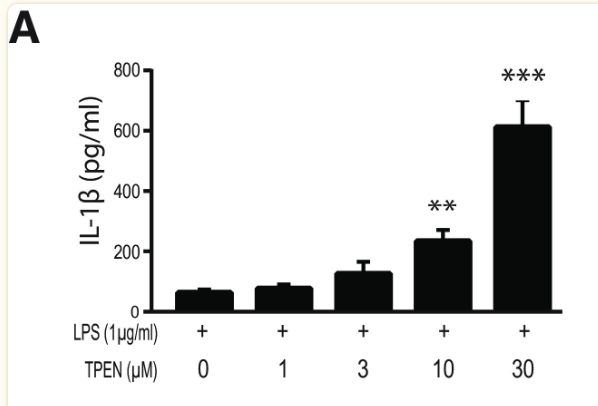
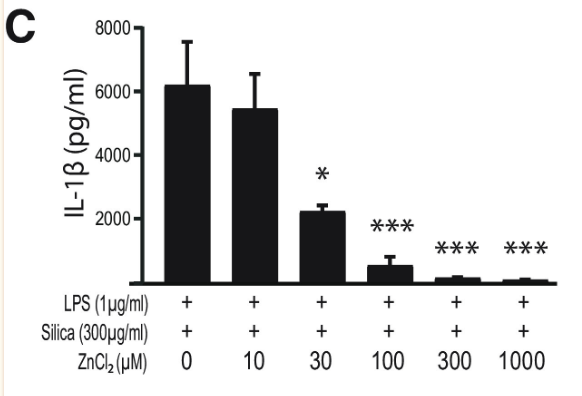
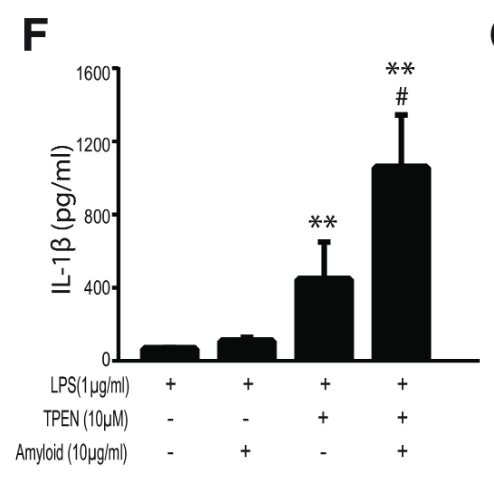
Finally, to assess the effect of zinc concentration on NLRP3 activity, NLRP3 release of IL-1β was measured under varying concentrations of TPEN, a zinc chelator, and zinc chloride. Increased TPEN concentration resulted in an effective decrease in free zinc, and additionally resulted in greater release of IL-1β, demonstrating increased NLRP3 activity in zinc-deficient conditions (Figure 9). Similarly, increased zinc chloride resulted in decreased IL-1β release, suggesting an inhibitory role of zinc regarding the NLRP3 inflammasome (Figure 10). Finally, amyloid oligomers were found to increase IL-1β release in the absence of zinc, suggesting the potential crosstalk between NLRP3-dependent and Aβ plaque-dependent Alzheimer’s progression (Figure 11).
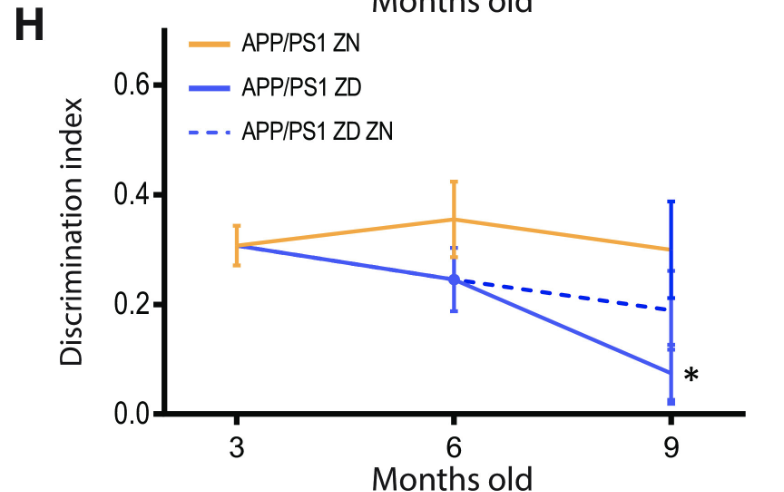
This research suggests a pathway through which zinc-deficient conditions may lead to excessive activation of the NLRP3 inflammasome, triggering Alzheimer’s Disease progression through exorbitant inflammation promoted by NLRP3 activity, notably independent from Aβ plaque formation. Additionally, should specific Alzheimer’s cases be identified as products of hyperactive NLRP3, this research suggests a potential therapeutic role of zinc supplements in slowing disease progression. Increased zinc chloride concentration resulted in decreased IL-1β production and therefore decreased NLRP3 activity, while ZD/ZN mice did not display statistically significant cognitive decline relative to ZN mice. These findings suggest a role of zinc supplements in inhibiting NLRP3 and thereby inhibiting the excessive inflammation that it may cause, while also pointing towards the ability of zinc supplements to slow cognitive decline despite three months of a zinc-deficient diet in the ZD/ZN mice.
Citations:
Xu, Y., Xiao, G., Liu, L. et al. Zinc transporters in Alzheimer’s disease. Mol Brain12, 106 (2019). https://doi.org/10.1186/s13041-019-0528-2
Rivers-Auty, J., Tapia, V. S., White, C. S., Daniels, M., Drinkall, S., Kennedy, P. T., Spence, H. G., Yu, S., Green, J. P., Hoyle, C., Cook, J., Bradley, A., Mather, A. E., Peters, R., Tzeng, T. C., Gordon, M. J., Beattie, J. H., Brough, D., & Lawrence, C. B. (2021). Zinc Status Alters Alzheimer’s Disease Progression through NLRP3-Dependent Inflammation. The Journal of neuroscience : the official journal of the Society for Neuroscience, 41(13), 3025–3038. https://doi.org/10.1523/JNEUROSCI.1980-20.2020
Zheng, H., & Koo, E. H. (2006). The amyloid precursor protein: beyond amyloid. Molecular neurodegeneration, 1, 5. https://doi.org/10.1186/1750-1326-1-5
Xudong Huang, Math P. Cuajungco, Craig S. Atwood, Robert D. Moir, Rudolph E. Tanzi, Ashley I. Bush, Alzheimer’s Disease, β-Amyloid Protein and Zinc, The Journal of Nutrition, Volume 130, Issue 5, May 2000, Pages 1488S–1492S, https://doi.org/10.1093/jn/130.5.1488S
Kelley, N., Jeltema, D., Duan, Y., & He, Y. (2019). The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. International journal of molecular sciences, 20(13), 3328. https://doi.org/10.3390/ijms20133328
Man, V. H., He, X., Ji, B., Liu, S., Xie, X. Q., & Wang, J. (2019). Molecular Mechanism and Kinetics of Amyloid-β42 Aggregate Formation: A Simulation Study. ACS chemical neuroscience, 10(11), 4643–4658. https://doi.org/10.1021/acschemneuro.9b00473
An, W.-L., Bjorkdahl, C., Liu, R., Cowburn, R. F., Winblad, B., & Pei, J.-J. (2005). Mechanism of zinc-induced phosphorylation of P70 S6 kinase and glycogen synthase kinase 3β in SH-SY5Y Neuroblastoma Cells. Journal of Neurochemistry, 92(5), 1104–1115. https://doi.org/10.1111/j.1471-4159.2004.02948.x