Copper is an important cofactor for the functioning of various cuproenzymes that are required for proper brain development and functioning. For example, in superoxide dismutase-1 (SOD1), the binding of copper is necessary to break superoxide radicals that exhibit neurotoxicity. Many cuproenzymes have chaperones, such as copper chaperone for SOD1 (CCS), to facilitate binding of the cofactor to the binding site. Failure to regulate copper levels in the body can result in various diseases such as Amyotrophic Lateral Sclerosis (ALS) and Huntington’s Disease (HD), which will be the focus of this post.
ALS is a neurodegenerative disease that affects nerve cells in the brain and spinal cord which leads to deterioration and loss of both upper and lower motor neurons. Loss of motor neurons weakens the muscles leading to paralysis and can be fatal if the muscles near the diaphragm are affected. ALS pathogenesis is caused by mutations in various genes such as C9orf72, SOD1, TAR DNA-binding protein 43, and RNA-binding protein FUS that lead to protein misfolding and aggregation. Mutations in SOD1 are commonly studied due to the formation of insoluble aggregates that exert neuron toxicity.
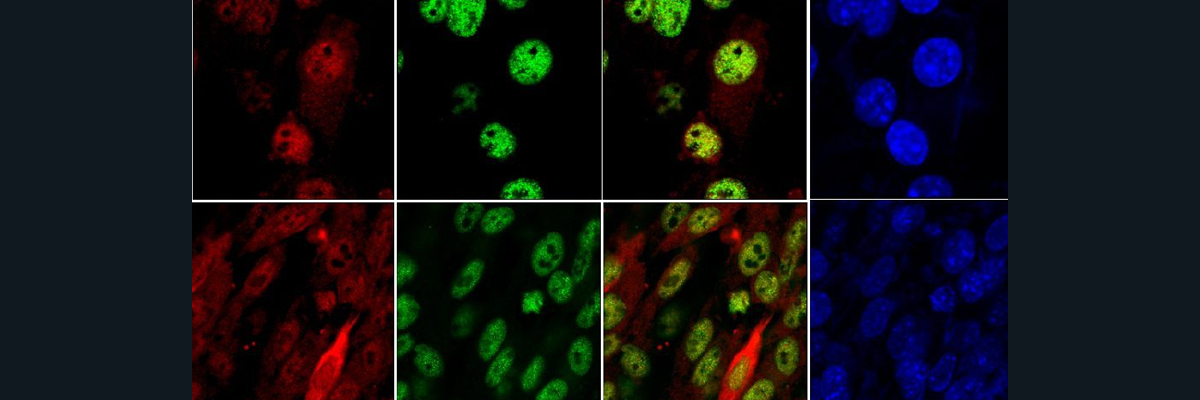
A study hypothesized that there is a correlation between metal deficient SOD1 proteins and aggregate formation. In SOD1, there are two metal cofactors, copper and zinc. Copper’s function in SOD1 is for catalytic activity whereas Zn serves for structural function as it strongly stabilizes and accelerates the kinetics of folding of the apoSOD. To test their hypothesis, they developed a novel antibody called anti-apoSOD to detect mutant SOD in ALS-model mice. The anti-apoSOD was able to recognize specific peptides that are not only aggregation prone but also contain cysteine and histidine residues that correspond to disulfide bond formation and bridging ligands for copper and zinc ions respectively. Through various experiments, they found that anti-apoSOD effectively detects mutant SOD1 that are disulfide bonded and are metal deficient at the metal binding site. It is specifically metal deficient and not necessarily copper deficient because it was found that zinc can bind at the copper-binding site of SOD1. The binding of zinc at the copper binding site decreased the effectiveness of anti-apoSOD recognizing the mutant SOD1. However, the signal intensity of the antibody recognizing mutant SOD1 proteins decreased over time, indicating the signal intensity is not correlated with the amount of SOD1 present. This reveals that high aggregation makes it difficult for anti-apoSOD to react and find the peptides that are aggregation prone. When anti-apoSOD was tested on human SOD1-ALS cases, the study found that the intensity of signal recognition was not as high. This is due to the tissue samples used for examination having an autopsy in the disease end-stage, whereas high intensity signals were prevalent in the early stages of the disease. Overall, this study provides evidence of anti-apoSOD being a potential diagnosis for determining early stages of ALS and leading to pharmacological intervention.
As of today, there are no permanent treatments for ALS. However, studies have shown that CuATSM is a promising treatment for ALS as it is known to deliver copper into the central nervous system to cells containing damaged mitochondria and directly to SOD proteins. A study found that although copper chaperone for SOD (CCS) is known to facilitate copper insertion in SOD1 to complete protein maturation, it also accelerates toxicity in mice co-expressing mutant SODG93A leading to early mortality. This is because overexpression of CCS has been hypothesized to impair copper important into mitochondria in low-expressing SODG93AxCCS. Therefore, scientists of this study hypothesized that by treating the mice with CuATSM can extend their life expectancy. After multiple experiments examining the effects of CuATSM on ALS-model mice, they found that earlier and continuous treatment of CuATSM reduces the percentage of copper deficient SOD, increases cytochrome c oxidase (a copper-containing metalloenzyme essential for ATP production) activity, and overall increases survival probability.
Huntington’s Disease (HD) is a neurodegenerative disease of the central nervous system caused by a mutation in the Huntingtin (HTT) gene. This mutation consists of extended repeats of the trinucleotide CAG which codes for the amino acid glutamine; an extended glutamine repeat of 36 or more will lead to HD pathogenesis. The HTT mutation is autosomal dominant, meaning that only one copy of the gene is necessary for development of the disease. Thus, each child with a parent with Huntington’s disease has a 50% chance of inheriting the disease and affects both men and women equally. Since HD initiates neuronal loss primarily in the striatum and cerebral cortex, some of the symptoms associated with HD changes in behavior, cognition, and physical impairment. Unfortunately, just like ALS, there is no current permanent treatment for HD, but there are temporary treatments to improve the quality of life of diagnosed patients.
A study investigated why increased copper levels promotes HD progression and they hypothesized that copper bound to low affinity sites could contribute to pro-oxidant activities and neurodegeneration. Scientists of this study investigated the affinity between Huntingtin gene fragments (N171-17Q) and copper (II) and iron (III). They found that the fragments have a higher affinity to copper (II) than iron (III), because in the Huntingtin sequence they found that histidine residues (82 and 98) were essential and specific to copper (II) coordination. Additionally, it was determined that the copper-N171 complex is redox-active as data indicated that copper (II) is reduced to copper (I) leading to aggregation of the Huntingtin protein. Certain metal chelators (Clioquinol and EDTA) were tested and determined to inhibit the formation of aggregates by decreasing redox activity of mutant Huntingtin protein. This is important because redox-active metal levels increase with HD progression as copper and iron concentrations in striata and frontal cortices are seen to increase with HD progression. However, the study determined that not all increased copper levels can be unassociated with the mutant Huntingtin.
Instead, it was found that the increased copper levels are associated with the inhibition of lactate dehydrogenase (LDH) activity, which are enzymes that play an important role in maintaining energy sufficiency for neurons. Therefore, the inhibition of LDH caused by increased copper levels is associated with neurodegeneration and progression of HD. Lastly, the concentrations APP, APLP1, and APLP2 (proteins associated with copper homeostasis) decreased with progression of HD, which confirms another reason for the high accumulation of copper. These findings encourage further investigation of copper interactions with mutant Huntingtin and LDH related to HD pathogenesis and can also lead to potential pharmacological interventions.
Fox, J. H., Kama, J. A., Lieberman, G., Chopra, R., Dorsey, K., Chopra, V., Volitakis, I., Cherny, R. A., Bush, A. I., & Hersch, S. (2007). Mechanisms of copper ion mediated Huntington’s disease progression. PLoS ONE, 2(3). https://doi.org/10.1371/journal.pone.0000334
Tokuda, E., Nomura, T., Ohara, S., Watanabe, S., Yamanaka, K., Morisaki, Y., Misawa, H., & Furukawa, Y. (2018). A copper-deficient form of mutant Cu/Zn-superoxide dismutase as an early pathological species in amyotrophic lateral sclerosis. Biochimica Et Biophysica Acta (BBA) – Molecular Basis of Disease, 1864(6), 2119–2130. https://doi.org/10.1016/j.bbadis.2018.03.015
Williams, J. R., Trias, E., Beilby, P. R., Lopez, N. I., Labut, E. M., Bradford, C. S., Roberts, B. R., McAllum, E. J., Crouch, P. J., Rhoads, T. W., Pereira, C., Son, M., Elliott, J. L., Franco, M. C., Estévez, A. G., Barbeito, L., & Beckman, J. S. (2016). Copper delivery to the CNS by CUATSM effectively treats motor neuron disease in Sodg93a mice co-expressing the copper-chaperone-for-sod. Neurobiology of Disease, 89, 1–9. https://doi.org/10.1016/j.nbd.2016.01.020