Copper serves as a signaling and regulatory molecule as well as a cofactor of important enzymes in the brain, and is thus required for proper brain development and functioning. Irregularities in copper metabolism can lead to several disorders, including Menkes disease and Wilson’s disease. Menkes disease is the result of genetic mutations altering ATP7A, a copper transporter protein, that leads to copper deficiency. On the flip side, Wilson’s disease results from a build-up of excess copper in the body caused by mutations in ATP7B, a similar copper transporter. Copper is an often essential cofactor for enzymes due to its ability to facilitate the transfer of electrons and its ability to bind and activate oxygen. Cytochrome C Oxidase, a complex found along the Electron Transport chain in the Mitochondria, contains Copper groups that accept and transfer electrons to oxygen during respiration. Superoxide dismutase 1 (SOD1), required for the conversion of dangerous oxygen radicals into hydrogen peroxide, is dependent on Copper to activate Cu/Zn binuclear sites. The loss of motor functions and cerebral atrophies are associated with the loss of SOD1 activity and mutations in SOD1 have been linked to ALS. CTR1 imports copper from interstitial fluid into the cells of the Central Nervous System (CNS). Cox11, CCS, Atox1, and possibly others are shuttle proteins or chaperones that transport copper to SOD1 in the cytosol or from the cytosol into the mitochondria, upon entry into the cell. Copper also enters the Atox1 pathway where ATP7A and ATP7B transfer copper into the lumen of the pathway to be exported out of the cells or incorporated into essential metabolic enzymes, like dopamine-β-hydroxylase (DBH). This review discusses the mechanisms by which the loss of copper homeostasis leads to the loss of catecholamine homeostasis.
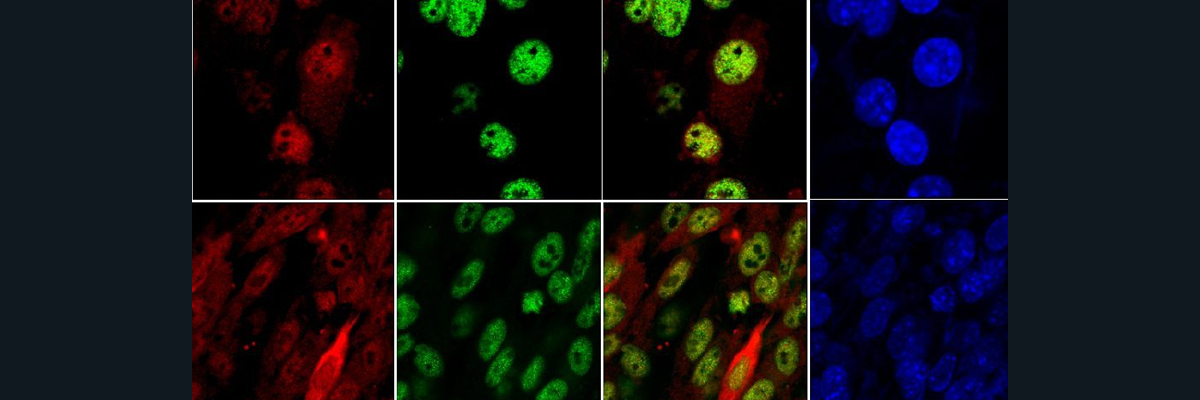
Catecholaminergic nerves include the dopaminergic and noradrenergic nerves. Dopaminergic nerves in the midbrain are responsible for posture, attention, and memory. Noradrenergic nerves are responsible for alertness and arousal through the neurotransmitter, norepinephrine. Copper is required for the conversion of dopamine into norepinephrine through the enzyme, dopamine-β-hydroxylase (DBH), that contains Copper binding sites. The loss of catecholamine homeostasis is seen in patients with Menkes and Wilkon’s Disease. In Menkes disease, where patients are deficient in copper, higher levels of dopamine and lower levels of norepinephrine in the brain are reported. This change in dopamine and norepinephrine levels are thought to be caused by the loss of copper incorporation into DBH, hence its decreased activity. In mouse models, where mice are deficient in copper (due to Menkes Disease), when the gene expression of ATP7A is restored and supplemental copper injections are administered, the regular levels of dopamine and norepinephrine are restored. Rats given copper deficient diets produced offspring with dopamine and norepinephrine imbalances. Patients with Wilson’s disease (excess build-up of copper) show variable changes in dopamine and norepinephrine concentrations, but some present dopaminergic system issues, although dopamine metabolism has yet to be properly mapped out. However, the accumulation of excess copper in the brain and body, as seen in Wilson’s disease, has been linked to mental illnesses like depression, psychosis, and sleep abnormalities. The area in the brain responsible for catecholamine metabolism, the Locus coeruleus (LC), is extremely rich in copper, as seen Figure 1 that shows an X-ray fluorescence imaging of the coronal section of a mouse brain that includes ventricle and locus coeruleus. You can see much more copper than phosphate in the LC region and in the ventricle as indicated by the lighter color that indicates higher concentration. Noradrenergic nerves are found in the LC, the body’s major supplier of norepinephrine. In figure 4, a mouse brain underwent immunostaining for DBH and the LC is seen to be rich in green fluorescence which indicates high concentrations of DBH that is established as a marker of noradrenergic neurons. Neurons in the LC, along with having high productions of proteins (like DBH) that are involved in catecholamine metabolism, also express a lot of CTR1 which contributes to it’s high copper concentration as seen in Figure 1. Zebrafish, rats, and humans have all been found to exhibit this high LC copper concentration that suggests it is evolutionarily conserved. Figure 5, an XFM analysis shows high amounts of copper deposits in the noradrenergic neurons in the LC’s of adult mice, but in humans, the Cu levels in noradrenergic neurons are uneven. Parkinson’s disease patients report decreased levels of neurons, copper, and CTR1 in the LC. Further studies must be done to establish a correlation between changes in copper concentrations and loss of neurons in neurodegenerative disorders. In conclusion, experimental and clinical evidence suggests an essential role of copper homeostasis in the CNS. The LC, where most of the body’s norepinephrine is produced, has been identified as a copper rich region where copper-dependent enzyme, DBH, has high activity. Further investigation may clue us into the other roles of copper in the LC and in catecholamine metabolism.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6941745/pdf/nihms-1541861.pdf