Iron is an important transition element and it is essential for several biological processes due to its inherent redox properties, like oxygen and electron transport, and are required by several enzymes. Iron is present in the circulation bound to the plasma iron carrier protein transferrin. Under iron overload conditions iron can also exist in the form of non-transferrin bound iron. Although iron is a very important metal for metabolism however if homeostasis is not maintained can cause serious implications like excess deposition of iron can lead to the generation of reactive oxygen species by Fenton reaction which produces superoxide and leads to the formation of hydroxyl radical which causes DNA damage and can cause tissue fibrogenesis.
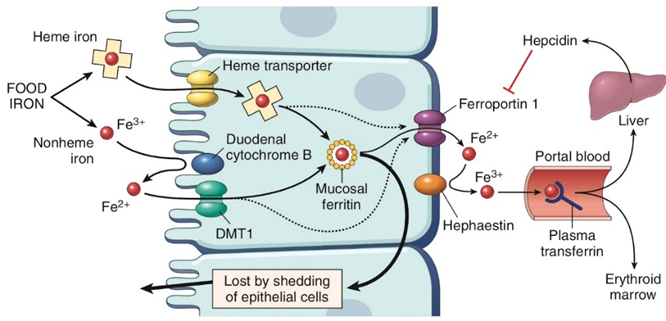
Iron through food sources gets absorbed in the small intestine. In the duodenum enterocyte, the iron comes in Fe3+ (ferric) but cannot be absorbed in Fe3+ form so it is reduced to Fe2+ form by the cytochrome B which has the enzyme ferroreductase. Then it is imported by the divalent metal transporter channel. Inside the cell, the iron gets converted back to ferric form for storage. Iron leaves the cell through the basal surface by a transporter Ferroportin. Iron is then again converted to ferric form by the enzyme hephaestin to be transported around. In the blood vessels, the protein transferrin (TFR) binds to iron to circulate throughout the body (Fig. 1). The majority of the iron goes to the bone marrow for erythropoiesis and the other part of iron gets transported to the liver where the iron is stored in ferric form as ferritin and can be released into the circulation through ferroportin when required. This iron homeostasis is maintained by hepcidin which is the major regulator that coordinates the plasma iron levels by binding to the exporter ferroportin which mediates the degradation of ferroportin and inhibits iron transfer into the circulation.
Iron though an essential micronutrient is potentially toxic at excess levels and leads to iron overload diseases like hemochromatosis, aceruloplasminemia, Genetic anemias with iron overload, Neuroferritinopathy, Transfusion-related iron overload, etc.
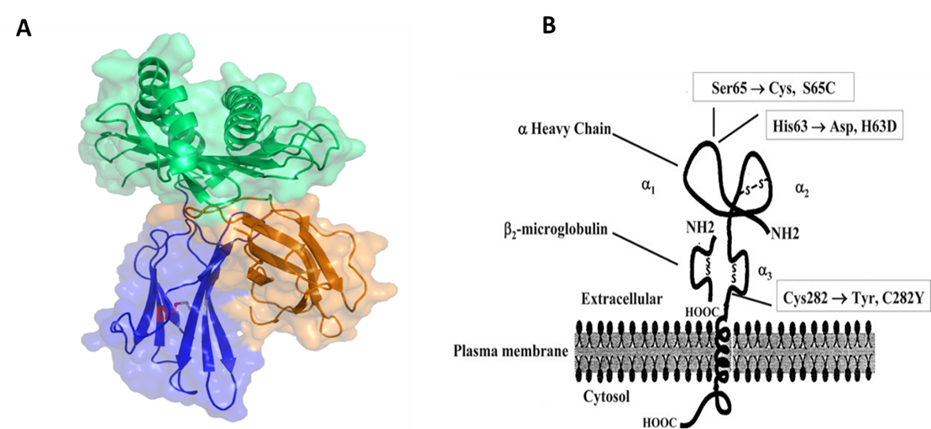
Hemochromatosis is inherited by autosomal recessive transmission it is mapped to the short arm of chromosome 6 close to the HLA-A locus and has two mutations in the HFE gene. The most predominant mutation in the gene is the substitution of tyrosine residue for cysteine residue at the 282 position (Fig. 2). The MHC class 1 family of molecules has 37% sequence identity and so it was proposed that the predominant mutation C282Y prevents the formation of a crucial disulfide bridge and as a consequence has an important effect on HFE protein structure. Thereby it causes an impact on the function and leads to iron overload disorder. Studies have revealed that C282Y /H63D compound heterozygous genotype is accounted for most hemochromatosis cases. While there is another mutation the H63D amino acid change has little effect on protein structure and does not produce a significant loss of function. This variant requires the presence of additional risk factors such as trans C282Y mutation or excessive alcohol intake.
Hemochromatosis can be classified into two types based on hereditary or not. Primary hemochromatosis is due to a mutation in the gene HFE which leads to low hepcidin on the other hand secondary hemochromatosis is characterized by ineffective erythropoiesis leading to increased duodenal uptake of iron, deposition of iron in the liver causing sideroblastic anemia and repeated transfusion can deliver a very large amount of iron which is accumulated in the organs. Hemochromatosis is more prevalent in males than in females as in females because there is the natural loss of iron through menstrual bleeding. Diagnosis is by liver LFT, biopsies, iron profiling also genetic testing for the presence of the mutation. Treatment involves phlebotomy and iron chelation. The clinical implications involved iron absorption in the intestine are increased and overburdening the cells causing free radical injury and organ damage.
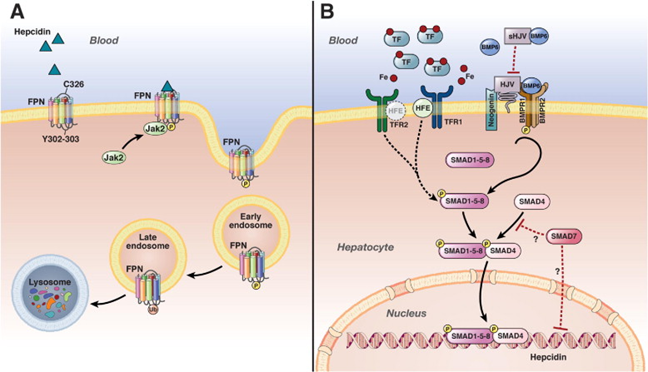
Hepcidin is the major regulator of dietary iron absorption, maintains the iron levels by preventing the release of iron into circulation, and also inhibits the systemic release of iron from the recycled enterocytes. Circulating iron bound to transferrin is sensed by TFR2 and HFE to regulate hepcidin production. The Hepcidin-ferroportin axis plays a critical role in regulating systemic iron homeostasis (Fig. 3). The iron content of the blood is monitored by the hepatocytes. BMP ligands initiate a signal transduction cascade. The process involves the phosphorylation of intracellular receptor-activated Smad1,5,8 which interact with Smad4 in the cytoplasm. The resulting complex then translocates to the nucleus, where it activates the transcription of the hepcidin gene. There is also an ERK signaling pathway involved which suppresses SMAD 1/5/8 signaling.
In the paper, “New thiazoldinones reduce iron overload in mouse models of hereditary hemochromatosis and β-thalassemia” the authors have targeted hepcidin generation as a therapeutic approach for iron-overload disorders. They developed novel chemical derivatives Thiozoldinones and their efficacy in reducing the iron overload in the organs.
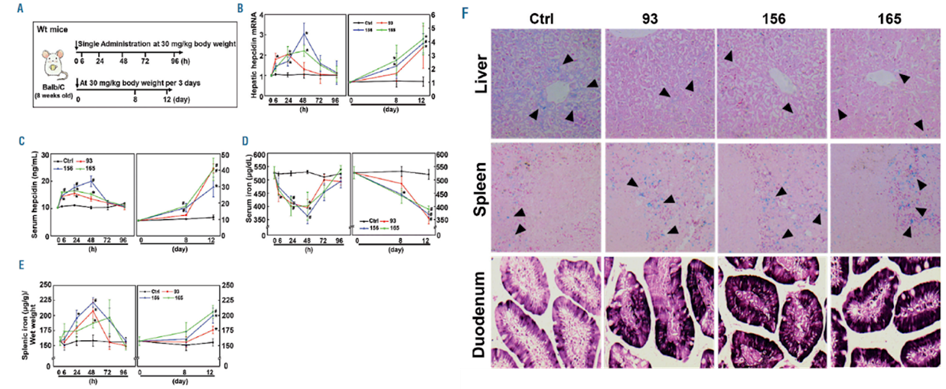
The authors have established a library of thiazolidinone derivatives and determined the lead compounds that can be used in hit-follow-up approaches for discovering novel thiazolidinone-related compounds. They designed a thiazolidinone compound library that is a Hepcidin agonist by incorporating several R1 and R2 groups on the thiazolidinone core. They created a combinatorial library of 210 compounds. Then they performed a high throughput screening of the thiazoldinone library by using a dual-luciferase reporter assay. For determining the hepcidin stimulating activity of the potential agonists in vivo they administered them by intraperitoneal injection of wildtype Balb/C mice at a dose of 30 mg/kg body weight. And could see that compounds 93 and 156 and 165 significantly increased hepatic hepcidin mRNA expression.
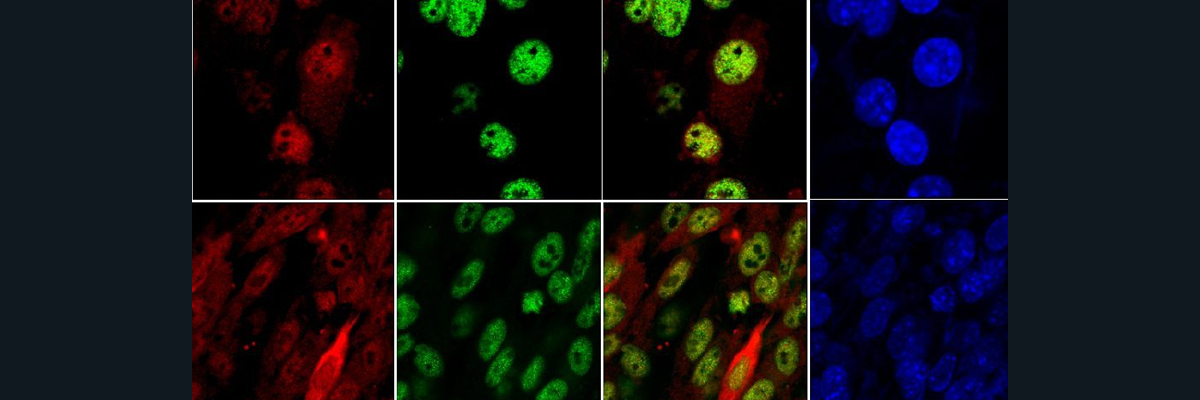
Upon administration of the selected compounds in the mice, there was a significant increase in the hepcidin expression which consecutively decreased the iron levels in the circulation and deposition in the organs. They validated these results by the liver and spleen iron staining (Fig. 4). which showed increased iron accumulation in the duodenum of mice treated with the compounds suggesting inhibition of iron transfer from the intestine to plasma due to increased hepcidin driven by these compounds. Also, they saw a reduction in transferrin saturation and serum ferritin levels which indicates less iron in circulation.
They tested the efficiency of these compounds in thalassemia intermedia, which they performed in Hbbth3/+ mice. They observed that the serum hepcidin was significantly increased by ~45% in Hbbth3/+ mice treated with compounds 93, 156, and 165, relative to the levels in untreated mice as seen in (Fig. 5). Since the iron overload anemia has damaged erythropoiesis, they saw that the compounds increased the number of red blood cells with normal morphology and a decreased damaged or deformed erythrocytes in treated mice which they confirmed with flow cytometric assay.
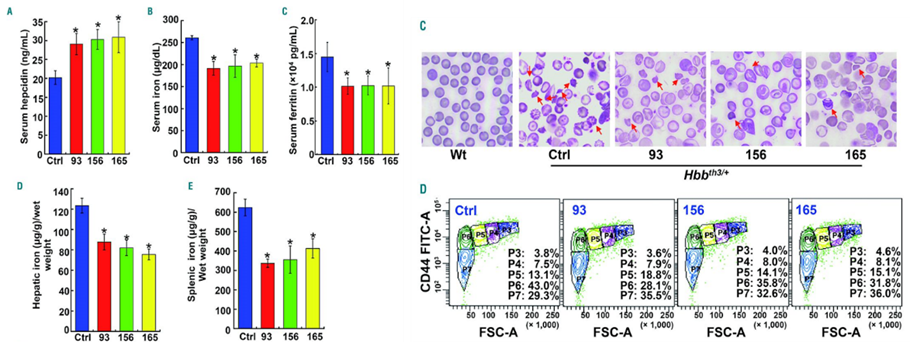
They concluded from their data that thiazolidinone derivatives 93, 165, and 156 can be used as therapeutically important hepcidin stimulatory activity, and the stimulatory effect on hepcidin synthesis lasted for 3 to 4 days after a single dose in mice. Also, the three compounds greatly increased hepcidin concentration and relieved or prevented iron overload in two mouse models, Hfe knock out mice which is representative of type 1 HH) and β-thalassemia intermedia mice, and prevented excessive iron deposition in organs of young Hfe KO mice.
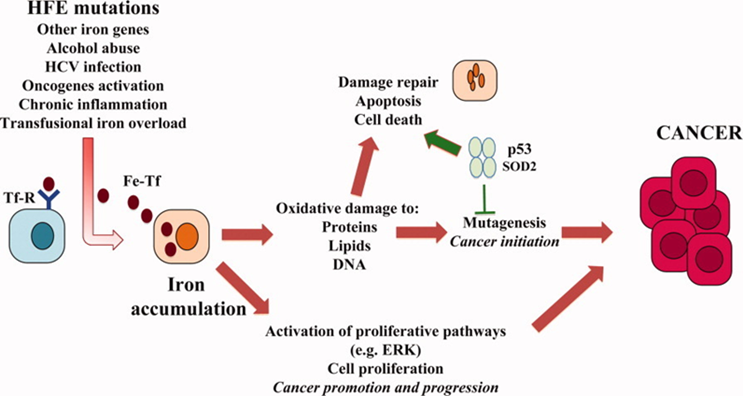
Hereditary hemochromatosis associated with iron deposition in the liver makes it more susceptible to hepatocellular carcinoma which is elucidated in the paper “Association between hereditary hemochromatosis and hepatocellular carcinoma (HCC)”
Iron accumulation can also lead to cirrhosis and the subsequent development of HCC, indirectly through the induction of chronic inflammation. Excess hepatic iron promotes the activation of hepatic stellate cells in HH which can promote fibrogenesis. The combination of elevated iron levels with environmental factors such as excessive alcohol consumption, viral hepatitis, and steatosis may act synergistically and lead to the development of HCC. Several studies showed Iron has a direct effect on tumor growth by promoting cellular proliferation. In human HCC cell lines, iron enhances proliferation and iron deprivation leads to cell cycle arrest and increased apoptosis. Interestingly, iron reduces immune surveillance for malignant transformation by impairing T-cell proliferation and inhibiting the tumoricidal activity of macrophages. Epigenetic alterations due to iron overload have also been implicated in hepatocarcinogenesis. Epigenetic defects such as increased DNA methylation commonly occur in HCC
References:
- Liu J, Liu W, Liu Y, Miao Y, Guo Y, Song H, Wang F, Zhou H, Ganz T, Yan B, Liu S. New thiazolidinones reduce iron overload in mouse models of hereditary hemochromatosis and β-thalassemia. Haematologica. 2019 Sep;104(9):1768-1781.
- Ulvik RJ. The liver in haemochromatosis. J Trace Elem Med Biol. 2015;31:219-24.
- Corradini E, Buzzetti E, Pietrangelo A. Genetic iron overload disorders. Mol Aspects Med. 2020 Oct;75:100896.
- Camaschella C, Nai A, Silvestri L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica. 2020 Jan 31;105(2):260-272.
- Le Gac G, Férec C. The molecular genetics of haemochromatosis. Eur J Hum Genet. 2005 Nov;13(11):1172-85
- Xu Y, Alfaro-Magallanes VM, Babitt JL. Physiological and pathophysiological mechanisms of hepcidin regulation: clinical implications for iron disorders. Br J Haematol. 2021 Jun;193(5):882-893
- Gattermann N. (2009). The treatment of secondary hemochromatosis. Deutsches Arzteblatt international, 106(30), 499–I.