Note: I highly recommend you read “Discovery of an Atomic Structure of Bacterial Cu+-Transport Protein Reveals Insight About Menkes/Wilson Disease” before this blog for clarification and background.
In my previous blog, I talked about the solved atomic structure of Legionella pneumophilia CopA (LpCopA) and how the solved structure offered insight on the molecular pathway of copper exiting the cytoplasm and into the periplasmic space. We also focused how the crystal structure allowed for illustrating Menkes/Wilson mutations, revealing that core residues in the binding domains correspond to greater severity of the genetic conditions. For this blog, I wanted to focus more on the putative Cu+-transport mechanism and how that can be further studied to understand the molecular mechanism behind the bacterial Cu+-ATPase.
Crystal structures offer great information on the static structure of a protein; however, it does not reveal any temporal detail of the structure nor the dynamics of the surrounding environment (i.e., ion-ion interactions, lipid-membrane motion, etc.). In an effort to understand the dynamic interactions of LpCopA, researchers Christina Gronberg and colleagues utilize computational simulations as a primary means of modeling the structural dynamics between the protein and external stimuli. It is noteworthy that the authors recognize the limitations of fully parameterizing the molecular environment (limited by complexity of real-life molecular dynamics and technological capabilities), and therefore supplemented their simulations with experimental testing.
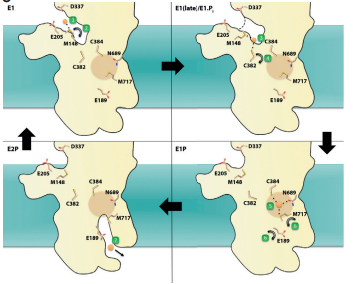
Gronberg and colleagues extensively sampled 150ns trajectories of lipid bilayer interacting with the TM domain. They identified several positively charged lysines and arginines that are involved in anchoring TM domain into the cellular membrane. Next, they sampled the docking platform (MB’ helix) and made note of the frequent contacts of the positively charged residues with lipid phosphates/oxygens and water solvent. What they found in their simulations suggested the importance of copper binding to K135, R136, and K142 and their point mutation (substituting an amino acid with another one) experiments confirms the necessity of these positively charged amino acids for Cu+-ATPase activity.
Subsequently, the researchers observed two key residues in their simulation of copper traveling through the TM domain: cysteine 382 and methionine 148. Previously, both residues were proposed to have a role in the ion-transport mechanism; both the MD simulations and experimental mutations validate this idea. Other residues that were identified in this study showed later on C384, N689 and M717 were vital in enabling copper to navigate, otherwise the function of the ATPase seems to be drastically be reduced for in vivo and in vitro testing.
Much of these residues are mostly identical to the proposed mechanism in Gourdon et al (2011), however a subtle difference highlighted here is the order in which copper interacts with polarly charged residues.
References:
(1) Grønberg, C.; Sitsel, O.; Lindahl, E.; Gourdon, P.; Andersson, M. Membrane Anchoring and Ion-Entry Dynamics in P-Type ATPase Copper Transport. Biophysical Journal2016, 111 (11), 2417–2429. https://doi.org/10.1016/j.bpj.2016.10.020.