Hard metal lung disease (HMLD) is a rare illness caused by chronic exposure to cobalt and tungsten carbide. HMLD is most commonly developed by people who work in industries that use heavy metal tools that are ground down, heated, or both, releasing heavy metal particles into the air which can be inhaled. Cobalt is a hypoxia mimic which results in the lung epithelium responding as if there was a lack of oxygen, regardless of how much oxygen is present. Cobalt simulates hypoxia by inhibiting PHD. PHD is responsible for the repression of hypoxia inducible factors (HIFs). In the presence of oxygen, PHD modifies key residues of HIFα, resulting in degradation. Under hypoxic conditions there are insufficient amounts of oxygen for PHD to tag the HIFα subunits. The stabilized HIFα subunits translocate to the nucleus, complex with HIFβ (also known as ARNT) and together work as a transcription factor for a large array of genes. Interestingly, only a very small percentage of people who work in these high risk industries develop HMLD. This indicates that HMLD may have a genetic basis. HMLD has no well established treatments. Gaining a better understanding of how cobalt and other hard metals cause HMLD can help develop more effective treatments.
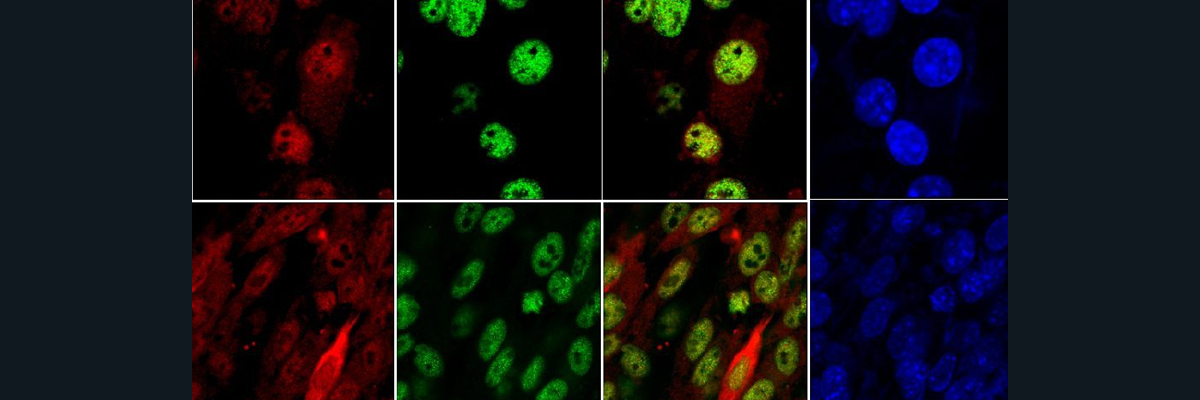
To investigate the mechanism behind HMLD, researchers use a DOX-inducible Cre recombinase system to eliminate HIF2α from mice postnatally. On day 4 a DOX (doxycycline) diet is started with the mothers of the DOX mice; this is continued until the mice are weaned on day 21. The DOX mice continue the DOX diet on their own until day 30. Researchers verified that the DOX-inducible Cre recombinase system eliminated HIF2α genetically using PCR, and phenotypically through immunohistochemistry (IHC). To investigate the potential role of HIF2α in cobalt-induced hypoxia, control and HIF2α∆/∆ mice were exposed to 1, 5, or 10 doses of either saline control or cobalt vehicle aspirations. A bronchoalveolar lavage (BAL) experiment was performed to obtain an immune response profile for each of the control and experimental conditions. While the control vehicle elicited little to no immune response, a general trend was observed under the cobalt treated mice. There was little to no immune response noted after 1 dose of cobalt, a large immune response amassed after 5 doses, and repression of the immune response after 10 doses. The outlier that failed to follow this trend was the concentration of eosinophils found in the HIF2α∆/∆ mice treated with cobalt. The trend holds true for 1 and 5 doses of cobalt, but instead of seeing a suppression at 10 doses, there was a significant escalation. This phenotype was verified using hematoxylin and eosin (H & E) stain and histopathology scores. Both bronchiolar and alveolar epithelium histopathology was investigated; the lack of suppression was only observed in the bronchiolar epithelium. To clarify eosinophil infiltration, IHC was performed on MBP, a cell-specific cytoplasmic constituent in eosinophil secretory granules. The trend held firm, confirming the previous results found. Finally, to gain understanding if these trends are unique to HIF2α or can be applied to the PAS family of transcription factors (the family that HIFs belong to) an AB-PAS stain was performed. The trend of the control developing an immune response after 5 doses of cobalt and suppressing the immune response by 10 doses was confirmed, as was the escalation of the immune response after 10 doses of cobalt for the HIF2α∆/∆ mice. The results of these experiments imply that HIF2α plays a significant role in the regulation of cobalt-induced hypoxia immune responses, especially in the regulation of eosinophils.