Of the metals we have been learning about in this course, Iron (Fe) is one of the most important metals that we all utilize in our everyday biological functions and our bodies need to keep a good balance in order for our bodies to function correctly. According to the World Health Organization (WHO), iron deficiency is the top nutritional disorder in the world. Research suggests that as many as 80 percent of people in the world don’t have enough iron in their bodies. It also suggests that as many as 30 percent of people have anemia due to prolonged iron deficiency[1].
Our dietary sources for iron are either in the heme form, which can be through eating anything that contains blood, or in the non-heme form (Fe3+) which can be ingested through various plants. Diets high in red meat, dark leafy vegetables, dried fruits and nuts, iron-fortified cereals, or bread can help treat or prevent iron deficiency. The recommended dietary allowance for Ages 19-50: Males- 8 mg Females- 18mg.
After Iron(Fe3+) is ingested, it is reduced to Fe2+ by a cytochrome on the surface of the enterocytes(cells that line our small intestine). After this reduction, the Divalent Metal Transporter 1(DMT1) is able to uptake the Fe2+ into the enterocyte. If the Fe2+ is in a free form, it comes into contact with ferroportin1 on the blood vessel side of the enterocyte which transports it out of the cell and into the blood (Ferroportin is the only known iron exporter in our cells). Our bones actually play a role in the regulation of iron by their production of transferrin, a protein which binds to the Fe2+ in the blood and transports it through the blood into the liver. When the Fe and transferrin are sensed at high levels, the hepatocytes from the liver releases hepcidin, which travels through the bloodstream back to the enterocytes and will inhibit the ferroportin, so that the transport of Fe is regulated. Hepcidin is able to limit iron export from the cell through its binding to ferroportin, which causes it to be internalized and later degraded. This is a preventative measure our body naturally does in order to not have too much Fe in our blood. However, the enterocytes do not stop intake of Fe through the DMT1 protein, and now are also unable to be transported out of the cell through ferroportin. Thankfully, our bodies are able to combat this by replacing the enterocytes every 3 days, and the iron that was not taken up in the blood is excreted by the body.
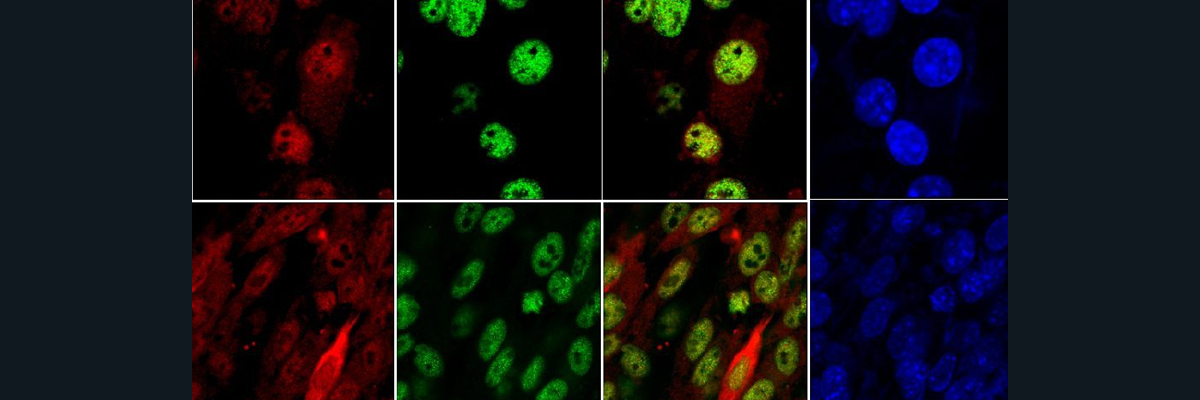
For the first experiment, they tested to see if a difference in iron diet would cause different expressions of hepcidin. They saw that the iron starved mice definitely had less hepcidin. Then, they also saw that the mice that were fed low iron showed HIF-1a expression, which shows that HIF-1a expression in the liver is primarily influenced by the dietary iron depletion. After breeding a strain that knocked out the HIF-1a gene entirely, another analysis of the hepcidin mRNA expression was taken and the experiment showed no significant difference between the hepcidin expression of the normal and the knockout mice.
Next, the group knocked out the VHL gene, which is thought to regulate the HIF-1 expression. The mice that were homozygous for the VHL deletion grew to only be half the weight of their normal wild type(WT) litter mates. Furthermore, the scientists calculated the percent weight that either the spleen or liver take up in the mutant and normal mice. We can see that the percentage is significantly larger for the VHL-/- (knockout) compared to the wild type (WT), which tells us that the deletion of VHL both caused the growth deficiency and the inflammation of the spleen and kidneys. The mutant mice also showed lower levels of total iron and ferritin levels compared to the WT. This makes sense, as a lower red blood cell (RBC) count correlates to low levels of total iron, which then is bound to ferritin inside our cells.
The following step this group did was to try to regulate the hepcidin levels by having HIF-1a bind to the promoter of hepcidin, which means that the binding of HIF made it so the mRNA levels of this protein will be reduced. Both the hepcidin mRNA and protein levels were significantly lower for the VHL-/- mice compared to the WT, indicating that VHL is involved in this iron pathway. A strain was then bred with an extra knockout of the ARNT gene, which stabilizes and activates HIF transcription. What they found was that the hepcidin decrease in VHL-/- mice is due to the stabilization of HIF-1a and HIF-2a, and not other function of the VHL gene.
This group ultimately found through these results that the cellular oxygen sensing and hypoxia-induced transcription are both largely mediated by HIFs. Another important discovery is that HIF may be one links that we have been looking for between iron homeostasis and hepcidin regulation, and HIF-1a has a direct repressor effect on the hepcidin promoter. However, deletion of the HIF-1 alone is insufficient in the compensation for hepcidin reduction. The largest takeaway from this article was that the VHL/HIF axis served a central role in coupling iron sensing to its regulation.[2]
[1] Miller, J. L. “Iron Deficiency Anemia: A Common and Curable Disease.” Cold Spring Harbor Perspectives in Medicine, vol. 3, no. 7, 2013, doi:10.1101/cshperspect.a011866.
[2] Peyssonnaux, Carole, et al. “Regulation of Iron Homeostasis by the Hypoxia-Inducible Transcription Factors (HIFs).” Journal of Clinical Investigation, vol. 117, no. 7, 2007, pp. 1926–1932., doi:10.1172/jci31370.