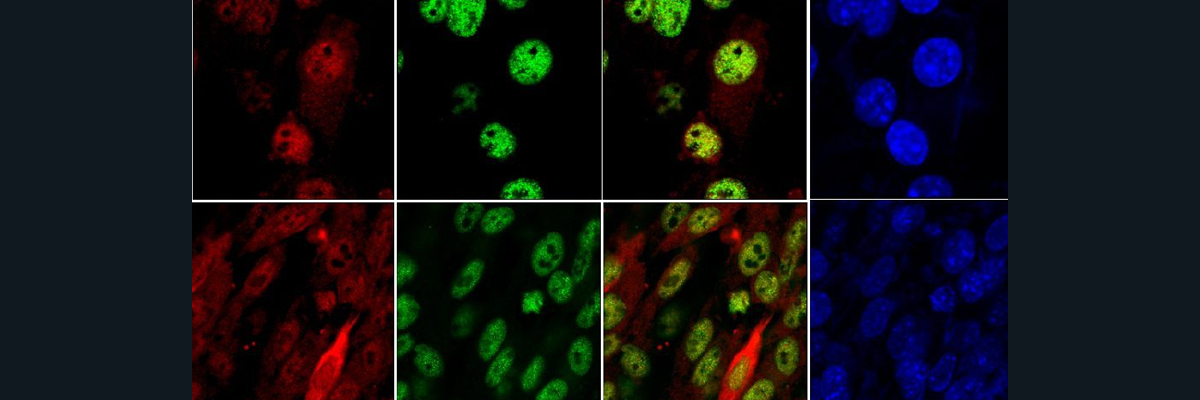
Parkinson’s Disease is a brain disorder which is caused by decreased levels of dopamine and the resulting degeneration of the neurons in the substantia nigra of the brain. Physical effects of Parkinson’s Disease include shaking, stiffness, issues with balance and coordination, memory loss and even dementia. As early as 175 AD, Parkinson’s disease has been documented and studied. Many advances have been made in the last century in the understanding of the causes of the disease. The current understanding of how the disease is caused is through the accumulation of structures called Lewy Bodies in neuronal cells. These structures are formed by the aggregation of alpha-synuclein protein fibrils. Once these fibrils are formed, a sequence of events occurs where membranous organelles including mitochondria become part of the Lewy Bodies. This complex formation is a major driver in the disruption of normal cell functions and the eventual neuronal cell death. To understand why the alpha-synuclein fibers form in the first place, researchers have investigated the relationship between alpha-synuclein and copper levels in the cell. It is well known that copper levels in the neuronal cells of Parkinson’s patients is significantly lower than in normal patients’ substantia nigra. Researchers have also discovered recently that the protein alpha-synuclein can interact with the copper chaperone molecule Atox1 and this interaction prevents the accumulation of the alpha-synuclein aggregates. It was further investigated through solution state NMR that there is a high binding affinity between copper carrying Atox1 and alpha-synuclein. The ssNMR data revealed that the binding between the molecules occurs at alpha-synuclein’s N-terminus near the known alpha-synuclein copper binding sites. Through surface plasmon resonance binding experiments, the equilibrium dissociation constant for acetylated alpha-synuclein (in vivo form of alpha-synuclein) to Cu-Atox1 shows extremely high affinity binding between the two molecules when Atox1 was loaded with Copper. Through fluorescence microscopy and atomic force microscopy, it was found that acetylated alpha-synuclein alone forms amyloid fibers, but in the presence of Cu-Atox1, the aggregation of acetylated alpha-synuclein fibers was prevented. When these amyloid fibers of acetylated alpha-synuclein were exposed to apo-Atox1 (no Cu), there was no reduction in the aggregation. This along with the ssNMR data confirmed that the prevention of the aggregation of amyloid fibers of alpha-synuclein was a copper dependent interaction. Further studies showed an interaction between Cu-Atox1 and acetylated alpha-synuclein in vivo, confirming that this interaction does take place in cells. More experiments need to be done to find a connection between copper metabolism and cellular dysfunction of Parkinson’s Disease, including investigating the function/dysfunction of the Cu-Atox1/alpha-synuclein complex and pathways to prevent Lewy Body formation.